Sample Preparation and Extraction #
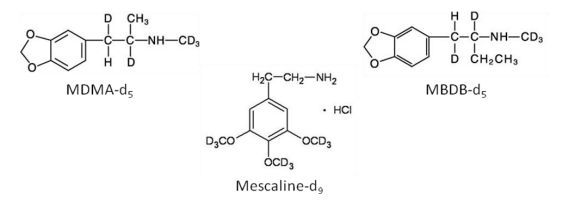
Sample Preparation and Extraction Methanolic working standards were used to prepare all calibrators and controls. Silanized glassware was used throughout. In the absence of deuterated analogs for each of the target drugs, a number of alternatives were evaluated. These included MDMA-d5, MBDB-d5 and mescaline-d9 (Figure 2). From a quantitative standpoint, mescaline-d9 yielded the most promising results during method development. This was not unexpected based upon its structural similarity (trimethoxy-derivative) to the target compounds (dimethoxy-derivatives). Internal standards used in other published methods are summarized in Table 4. In several instances, internal standards are not used, or bear very little structural similarity to the target analytes.
Urine sample preparation and extraction #
Drugs were extracted from urine by solid phase extraction (SPE) using PolyChrom Clin II mixed-mode polymeric columns from SPEware (Baldwin Park, CA). All extractions were performed using 2 mL of urine. Following the addition of mescaline–d9 internal standard to achieve a final concentration of 100 ng/mL, 2 mL of pH 6.0 phosphate buffer was added, samples were vortex mixed and transferred to SPE columns. Samples were drawn through the column under gravity, or sufficient vacuum to maintain continuous flow. Columns were then washed with 1 mL of deionized water, 1 mL of 1M acetic acid and then dried at full vacuum for 5 minutes. Columns were washed with hexane (1 mL), ethyl acetate (1 mL), and methanol (1 mL) in a successive fashion. Target analytes were eluted with 1 mL methylene chloride:isopropanol (95:5, v/v) containing 2% concentrated ammonium hydroxide. Acidic methanol (30 µL) was added to each sample prior to evaporation at 50oC using a TurboVap® II (Caliper Life Sciences, Hopkinton, MA). Extracts were reconstituted in 50 µL of mobile phase A and transferred to autosampler vials for LC/MS/MS analysis. For GC/MS analysis, extracts were reconstituted in 20 μL of ethyl acetate.
Analytical recovery was evaluated to determine the extraction efficiency of the SPE method. Additionally, the effect of “salting out” prior to evaporation was investigated to determine if sample losses could occur due to the volatility of some target drugs in the base (uncharged) form. Acidic methanol was used for this purpose as described above. Analytical recovery was determined by fortifying drug-free urine with target analytes at 100 ng/mL (N=5). The analytical recovery was estimated by comparison of the relative peak areas of target analytes. The extract containing internal standard (IS) alone was fortified with target drugs immediately after the extraction, prior to the evaporation step. Samples were reconstituted and analyzed by LC/MS/MS. The analytical recovery (extraction efficiency) was calculated from the relative peak area (drug/IS) of extracted and non-extracted samples.
Blood sample preparation and extraction #
Drugs were extracted from whole blood by solid phase extraction (SPE) using a modification of the procedure developed for urine. All extractions were performed using 1 mL of whole blood. Following the addition of mescaline–d9 internal standard to achieve a final concentration of 100 ng/mL, calibrators and quality controls were fortified with the appropriate volume of working standard. A variety of sample pretreatment steps and protein precipitants were evaluated, including acetonitrile, methanol, 10% sodium tungstate, 20:80 0.2 M zinc sulfate/methanol, 10% trichloroacetic acid (TCAA), acetone and 50% ethanol in 10% zinc sulfate. Simple dilution in phosphate buffer (0.1 M, pH 6) was also evaluated. During protein precipitation, 2 mL of the appropriate protein precipitation agent was added to the blood with vortex mixing. Acetonitrile was stored in the freezer prior to use. Samples were then centrifuged for 10 minutes at 8,000 rpm. Supernatants were diluted in pH 6.0 phosphate buffer (4 mL) and added to Cerex PolyChrom Clin II solid-phase extraction (SPE) columns from SPEware (Baldwin Park, CA). The SPE columns were attached to Teflon needle inserts and then a vacuum manifold, both from J.T. Baker (Phillipsburg, VA). Samples were drawn through the columns under gravity or with mild vacuum, as needed. The columns were washed with 1 mL of DIW and then 1 mL of 1 M acetic acid. Columns were dried for 5 minutes at full vacuum and then washed consecutively with 1 mL each of hexane, ethyl acetate and methanol. The designer amphetamines were then eluted with 1 mL of 2% concentrated ammonium hydroxide in 95:5 (v/v) methylene chloride: isopropanol. Acidic methanol (30 μL) was added to each extract after the elution step. The extracts were then evaporated using a Caliper Life Sciences TurboVap® II evaporator (Hopkinton, MA). Dried extracts were reconstituted in 50 μL of mobile phase A and 20 µL of the sample was injected onto the LC/MS/MS.
Isolation of Drugs #
Solid phase extraction (SPE) was used to develop and optimize a procedure for the extraction of fifteen psychedelic amphetamines from blood and urine. Samples were treated with acidic methanol prior to evaporation to minimize evaporative loss and imprecision. A statistical evaluation of analytes treated with acidic methanol prior to evaporation showed significant improvement compared with those that were not treated (and therefore evaporated in the base, or uncharged form). During the development and optimization procedure for whole blood, a variety of sample preparation techniques were evaluated. Acetonitrile protein precipitation and sample dilution in buffer were optimal. The latter was selected based upon analyte recovery, ease of use and analysis time. Analytical recoveries for all target compounds were 64-93% in urine and 60-91% in blood.
Note: Acidic methanol (1%) was prepared by adding concentrated hydrochloric acid to methanol in a ratio of 1:99 (v/v).
Liquid Chromatography/Tandem Mass Spectrometry #
Liquid Chromatography/Tandem Mass Spectrometry (LC/MS/MS) was successfully used to identify an expanded panel of psychedelic amphetamines in blood and urine.
Separation was achieved using a Shimadzu high-performance liquid chromatography (HPLC) system (Columbia, MD) with a Phenomenex Luna 5µm C18 Colum (100 x 2.0 mm) (Torrance, CA) equipped with a guard column (4.0 x 2.0 mm). An API 3200 tandem mass spectrometer from AB Sciex (Foster City, CA) and Analyst 1.4.2 software from Applied Biosystems were used for detection. Gradient elution was necessary for separation of the target analytes. Mobile phase A consisted of 50mM ammonium acetate in DIW/methanol (95:5). Mobile phase B consisted of 50mM ammonium acetate in a mixture of acetonitrile/DIW (90:10). A flow rate of 0.4 mL/min was used in accordance with the following gradient profile: 20% mobile phase B for 0-1 min, increased to 65% by 4 min, held at 65% until 4.5 min, then decreased to 20% by 6 min. Positive electrospray ionization (ESI) and multiple reaction monitoring (MRM) were used throughout. Reconstituted extracts were injected (30 µL) onto the LC/MS/MS using a Shimadzu Sil-20A HT autosampler equipped with two LC-20AT pumps.
Urine #
Limits of detection in urine were 0.5 ng/mL (2C-C, 2C-D, 2C-E, 2C-H, 2C-I, 2C-T-2, 2C-T-4, 2C-T-7, 4- MTA, DOB, DOC, DOET, DOI, DOM) and 1 ng/mL (2C-B). The limit of quantitation in urine for all drugs was 1 ng/mL. Accuracy for controls evaluated at 50 and 250 ng/mL were 96-120% for all target analytes and intra-assay CVs were 0.5-5.6% over the same concentration range. Interassay CVs in urine were 2.1-20.8% at 100 ng/mL. No matrix effects were observed and no interferences were present using structurally related compounds, endogenous bases and other common drugs in urine.
Blood #
In blood, limits of detection were 0.5 ng/mL (2C-D, 2C, E, 2C-H, 2C-I, 2C-T-2, 2C-T7, 4-MTA, DOB, DOC, DOET, DOI, DOM) and 1 ng/mL (2C-B, 2C-C, 2C-T-4). Limits of quantitation were 0.5 ng/mL (2C-D, 2C-E, DOI, DOM), 1 ng/mL (2C-B, 2C-C, 2C-H, 2C-I, 4-MTA, DOB, DOC, DOET) and 2 ng/mL (2C-T-2, 2C-T4, 2C-T-7). Whole blood controls at 50 and 250 ng/mL yielded accuracies of 89-112% and intra-assay CVs of 1.1-6.1%. Inter-assay CVs at 100 ng/mL were 2.9-8.2% for all target analytes. Following optimization of the procedure, no matrix effects were present using whole blood. In the interference study however, meperidine was responsible for a small but measurable suppression. As a result, two analytes (2C-T-2 and DOB) provided quantitative results lower than expected and just outside of the acceptable range (± 20%).
GC/MS #
Some published methods utilizing GC/MS derivatize these amphetamine-like drugs using acetic anhydride, n-butyric anhydride, isobutyric anhydride, heptafluorobutyric anhydride, and pentafluoropropionic anhydride. Derivatization has many advantages from the standpoint of improved detectability, volatility, specificity and chromatographic separation. However, in this study we found it unnecessary to derivatize the target analytes. Non derivatized drugs can be advantageous if a laboratory is making an identification using a commercial or widely used mass spectral library, particularly if the laboratory conducts full scan screening by GC/MS. The purpose of this study was to establish a simple procedure for the separation and identification of the target drugs, using techniques and instrumentation already widely used in human performance and medical examiner’s toxicology laboratories.
The injector and interface were both set at 280°C. Injections (2 μL) were made in split mode with a 5:1 split ratio. Ethyl acetate was used as the wash solvent, with a total of six pre-and post injection syringe washes between samples. The oven temperature was held at 130°C for 0.50 min, ramped to 170°C at a rate of 15°C/min with a hold time of 1 min, ramped to 180°C at a rate of 5°C/min with a hold time of 9 min, ramped to 200°C at a rate of 15°C/min and then ramped to 290°C at a rate of 30°C/min with a final hold time of 1 min. The total run time was 20.0 min. Helium was used as the carrier gas at a flow rate of 1.3 mL/min. The MS was operated in the electron impact (EI) ionization mode. The ion source and quadrupole were set at 230°C and 150°C, respectively.
Reference: US Department of Justice: Designer Amphetamines in Forensic Toxicology Casework, 2013