This is a transcript of the following video.
Okay, so now let’s look at the basic idea behind the classic theory of Raman scattering or Raman spectroscopy.
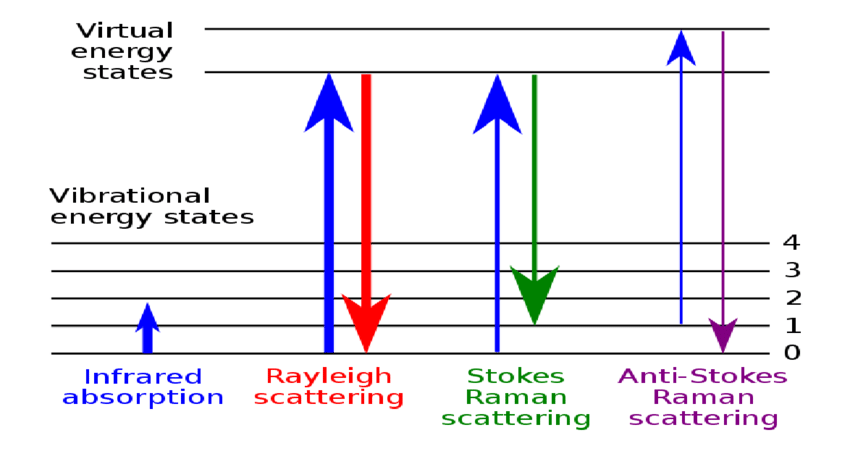
To begin with, and this is something we’re going to keep coming back to multiple times, as we talk about the spectroscopy, molecules 1 can exist in various energy levels. And we’re going to be talking about vibrations here. So this is going to be a vibrational energy level equals zero, or we’re going to call it the ground state. And then there can be various excited states, which we’ll just call v equals one, v equals two. And an infrared spectroscopy, we just take from v equals zero to v equals one, we absorb a photon. And there we are, we’re in the energy level.
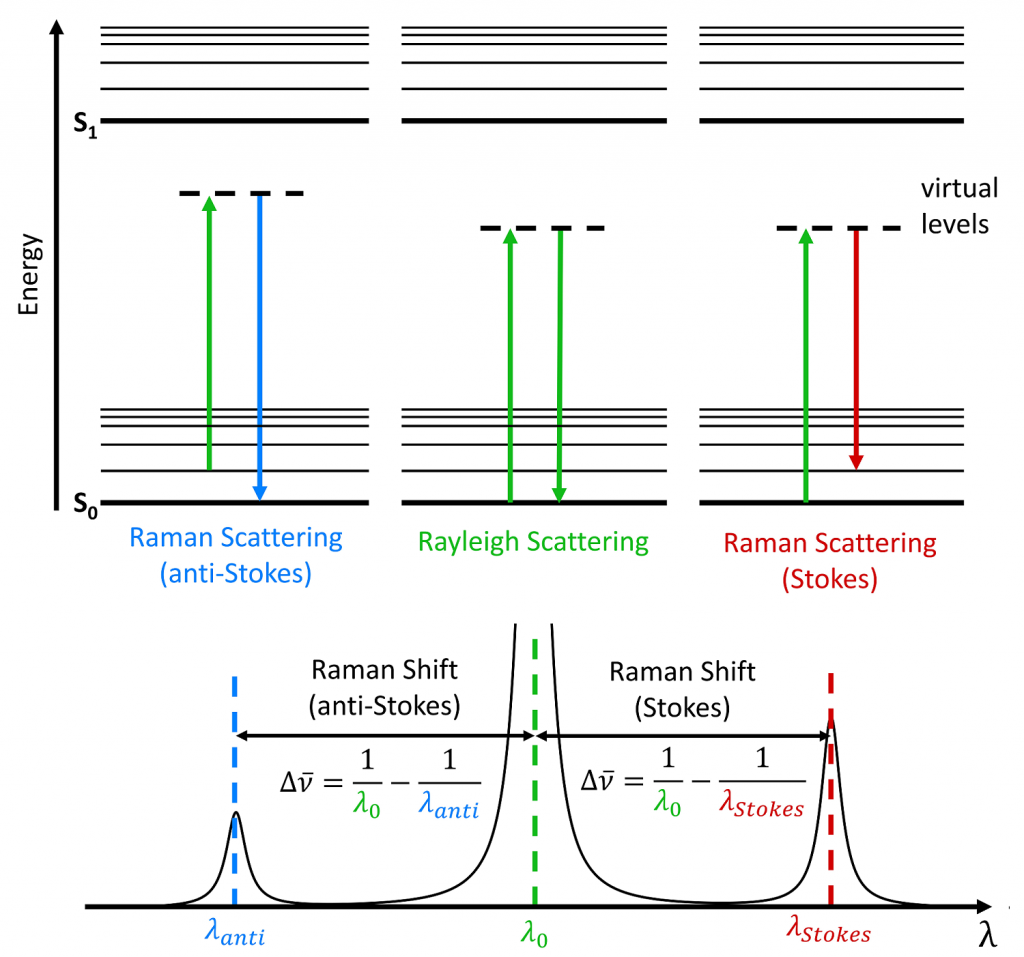
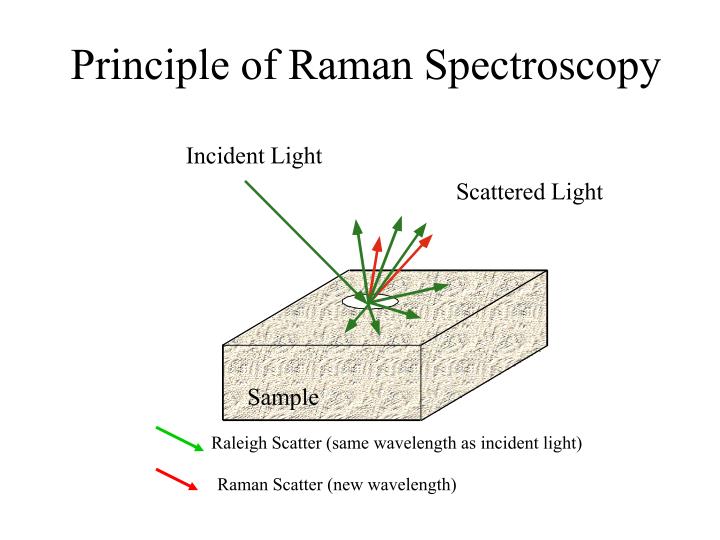
But in Raman, something a little more complicated happens. What happens in Raman is that we will shine a source on to the sample. In this case, this is going to be called a laser νL. And that laser frequency is going to when the molecule is struck by it, it’s going to excite that molecule up to something called a virtual state. Now that virtual state is not a real energy level, it’s just whatever energy that laser happens to have. And the molecule is raised up to that virtual state and it’s transient, it doesn’t stay there for very long. Now, once it’s there, one of two things can happen.
-
The first thing is it can scatter that light right back and go right back to the ground state. And this is going to be the νR. In this case, $$νR=νL$$. And the reason I call it R is because this is called Rayleight light, or Rayleight scattering. The Rayleight scattering comes out with the same energy that went in with. There’s no real energy interchange between the system here.
-
The other thing that can happen, though, is that the molecule can scatter the light, but while it’s scattering the light, or as that light is impinging on the virtual state, when the molecule is in the virtual state, it can take a little bit of the energy out, and the energy that it takes out gives it this excitation. This we’re going to call νS, and critically νS, which is called the Stokes line, and hence the S. $$νS = νL - ν0→1$$ 2 . What we do know, we know νL, that’s the laser, that’s our source, that’s the energy we put in. The spectrometer, as we’ll see in a later video, measures νS. It measures this scattered or Stokes photon. So we know these two. So if we solve this equation, we can find this energy. And that’s what we’re interested in. This is the energy of the molecule, and this is what the Raman is looking at.
Now, it is also possible for the molecule to initially start out in an excited state. Now that is νL again, it’s the same νL but since there’s a spacing here, it actually goes to a higher virtual state. And from that higher virtual state, the molecule can then scatter all the way back to the ground state. Notice in this case, and we’ll call this νAS, that this photon right here, which is called an anti Stokes photon, is higher in energy than the energy that came in, because the molecule gave up some energy. So if we look here, we have $$νAS = νL + ν0→1$$.
So you can see these are the three standard Raman events:
- The Rayleight scatter, in which case no energy is interchanged. The photon in, the photon out. As we’ll see, as we go on, this is an extremely intense photon, and actually can cause some problems, because it’s so bright, so intense, that it can actually damage the detector, if we don’t do something about it. We’ll see what we do about that later.
- The Stokes photon is going to be by far and away the most common that we’re going to look at where some of the energy has been removed from the laser. And that energy is equal to this vibrational excitation and
- the anti Stokes photon, which is hired energy, as we’ll see when we talk about out fluorescence, it has certain advantages, it would be nice to be able to use this. But because of something called the Boltzmann distribution, this is not always that useful.
So that’s the basic idea behind Raman. The three different scatters Rayleight Stokes and anti Stokes, the interchange of energy. And the key underlying fact, we are looking at a vibrational excitation, that vibrational excitation is the same thing that you’re looking at when you’re doing infrared spectroscopy, where you’d be going from this level to this level, it’s the same type of thing. As we’ll see, there’s different selection rules and different kinds of vibrations that we’re looking at. But nevertheless, it’s the same information. So we’re looking at that that excitation.