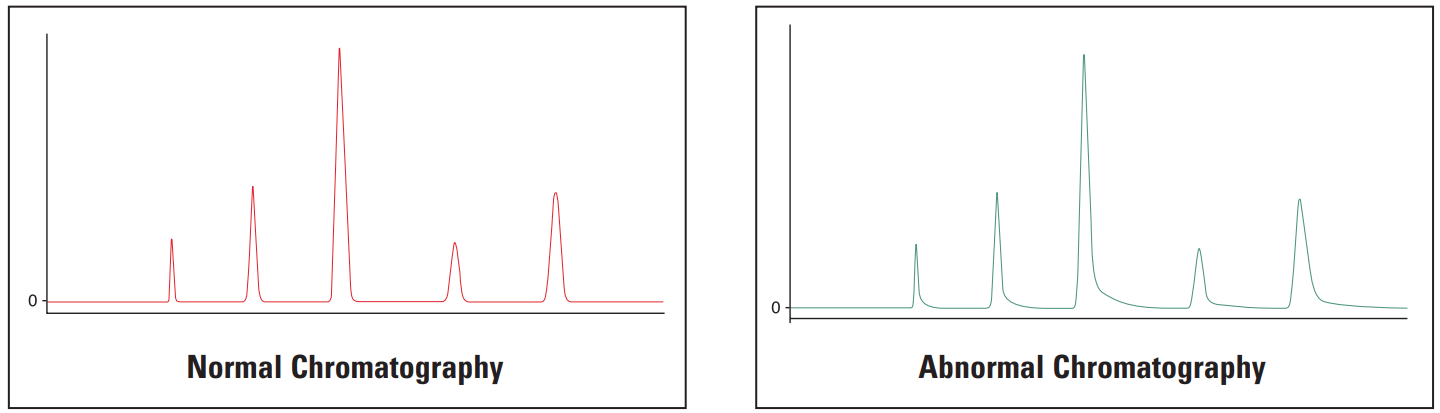
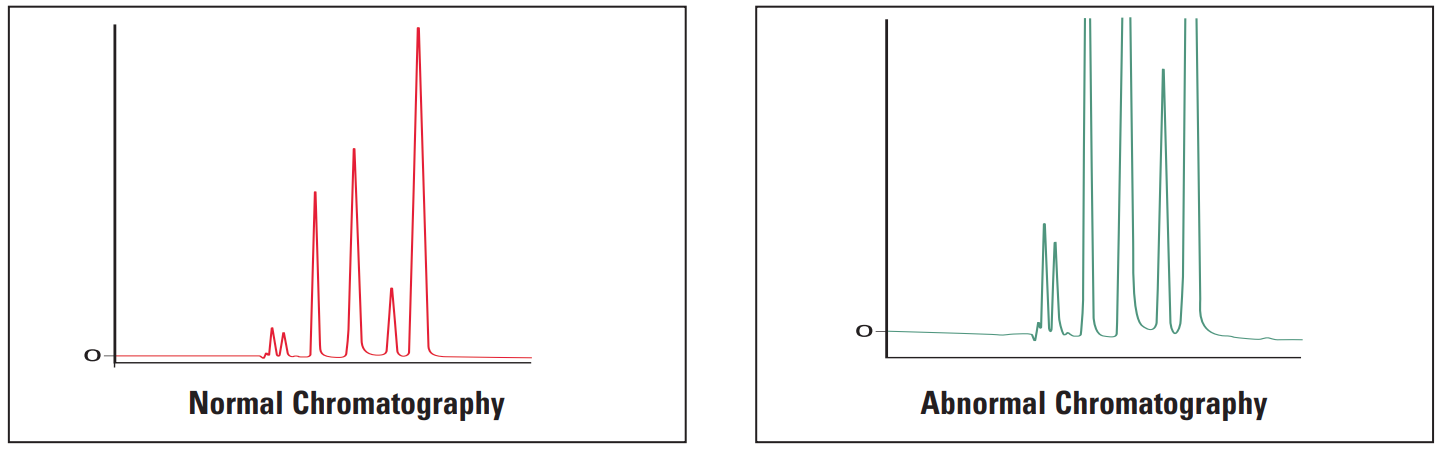
Abnormal peak shape encompasses a range of possible peak shape problems.
- No peaks
- Smaller than expected peaks
- Fronting or tailing peaks
- Broad peaks – early eluting analytes or all analytes
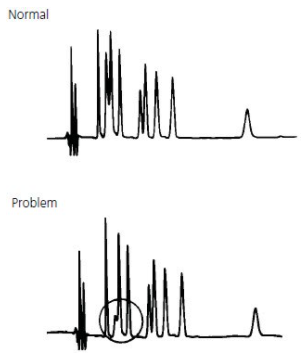
- Double peaks / shouldering peaks / Peak Splitting
- Flat topped peaks
- Change in Peak Height (one or more peaks)
- Rounded peaks
- Extra peaks
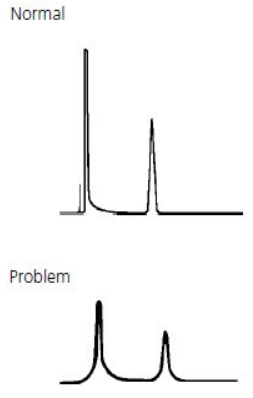
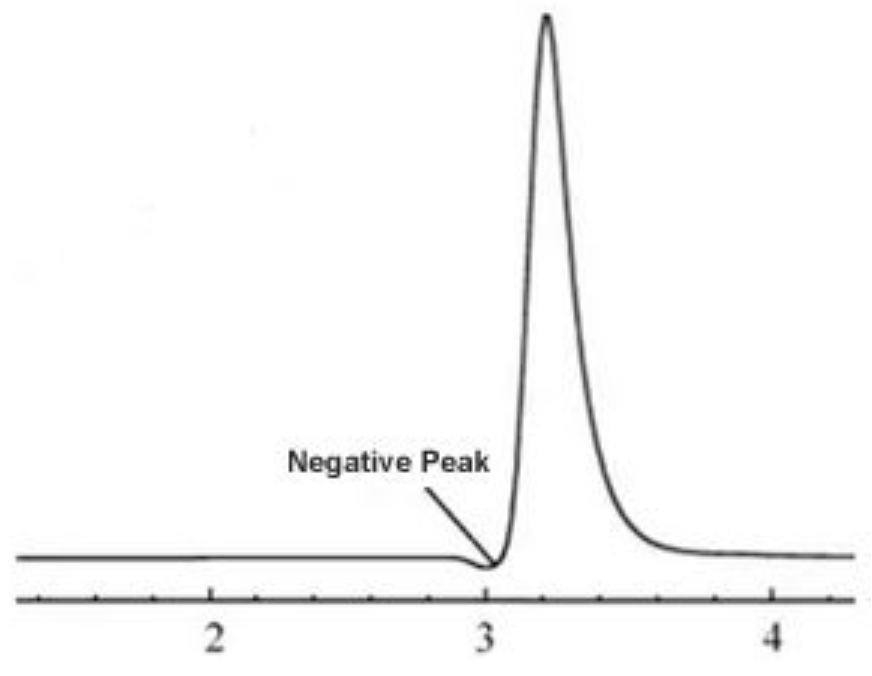
- Negative peaks
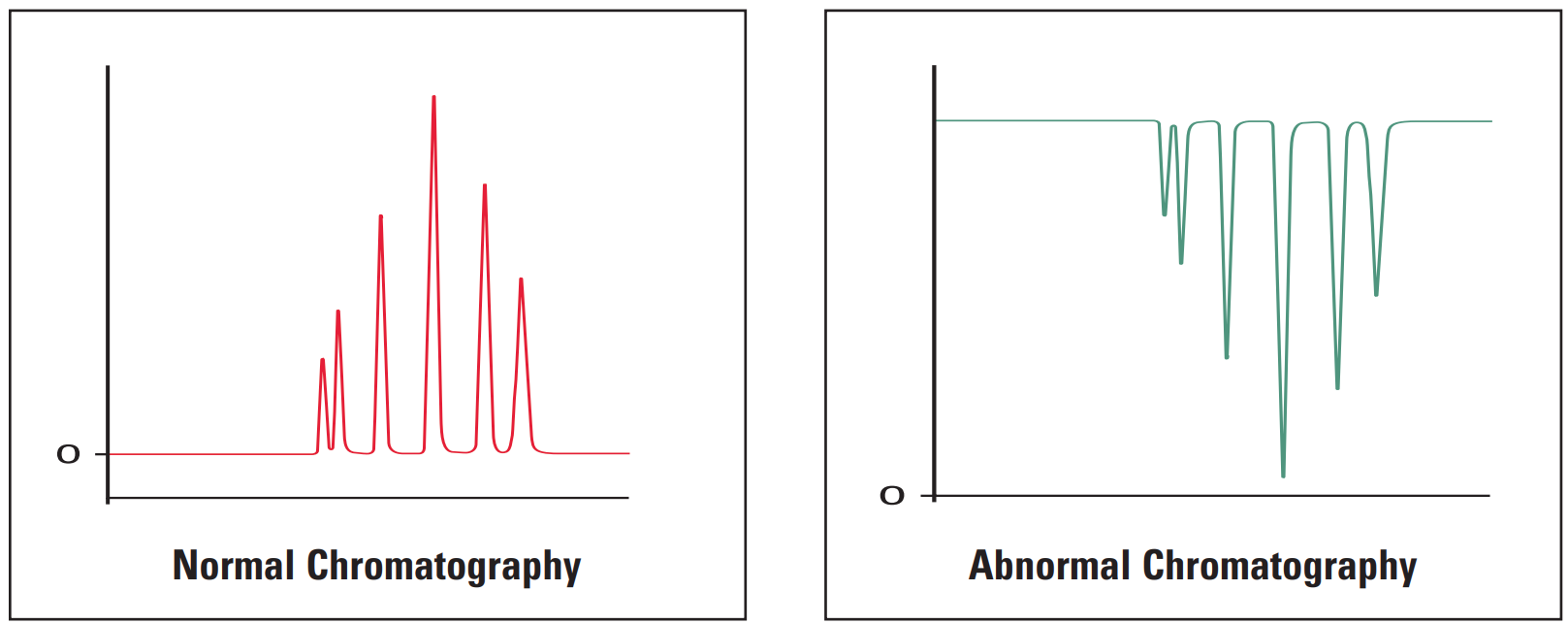
- Ghost peak
If all the peaks in a chromatogram are affected, then it suggests that the problem is related to either the system or the column. If only early eluting peaks are affected then it suggests that the problem lies within the fluid path – perhaps with incorrect ID tubing, fittings etc.
If single peaks are affected, then it suggests that there might be a specific chemistry problem. The method in use should be examined for areas where the chemistry may not be correct.
Gradient methods, where early eluting peaks are abnormal and later peaks are acceptable may be suffering from pre-column band broadening. If all the peaks are abnormal, then post-column band broadening or other changes in the system are the most likely causes.
Isocratic methods, where the early eluting peaks are abnormal and the later peaks are acceptable may be suffering from extra-column band broadening, injector problems, incorrect detector time constant or incorrect A/D sampling time. If all the peaks are abnormal, then extra-column band broadening or other changes to the system are the most likely causes.
Each table in this section will show an example of the type of chromatography being investigated; it’s cause(s) and any corrective action that can be taken.
No Peak / Missing Peaks #
Thermo Guide #
Lack of chromatogram peaks is often due to either the wrong sample being injected, the detector not being switched on or a blockage between the injector and detector lines.
The next most common reason for a lack of peaks is that some part of the sample or mobile phase preparation has been performed incorrectly, so it is always worth revisiting to check that the correct buffer has been used, the sample/solvent pH is correct etc.
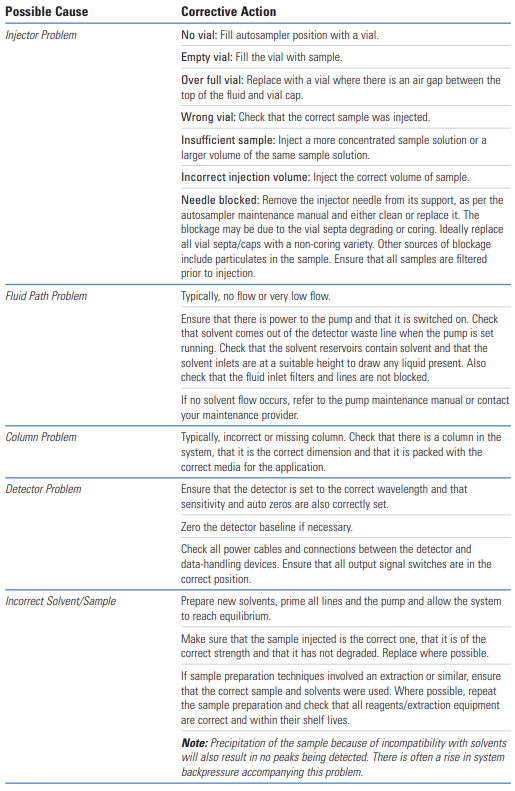
Single or multiple missing peaks are usually due to the wrong sample being injected or the sample degrading.
Equally likely though is a loss of resolution due to column/ solvent inconsistencies.
Sigma-Aldrich Guide #
Problem | Probable Cause | Remedy/Comments |
|---|---|---|
| 1. Detector lamp off. |
1. Turn lamp on. |
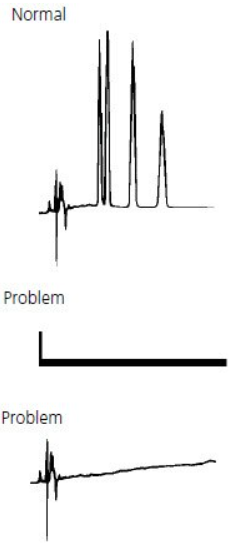
Smaller than expected peak #
Thermo Guide #
Smaller than expected peaks are often due to either the wrong sample being injected, an incorrect sample volume being injected, or a blockage between the syringe needle and detector. Problems with the syringe plunger sticking in the barrel can occur if the sample contains particulates.
Note: Viscous samples will require a longer draw time. Insufficient draw time will result in a lower volume of sample being injected onto the column and smaller peaks will result.
Galak Guide #
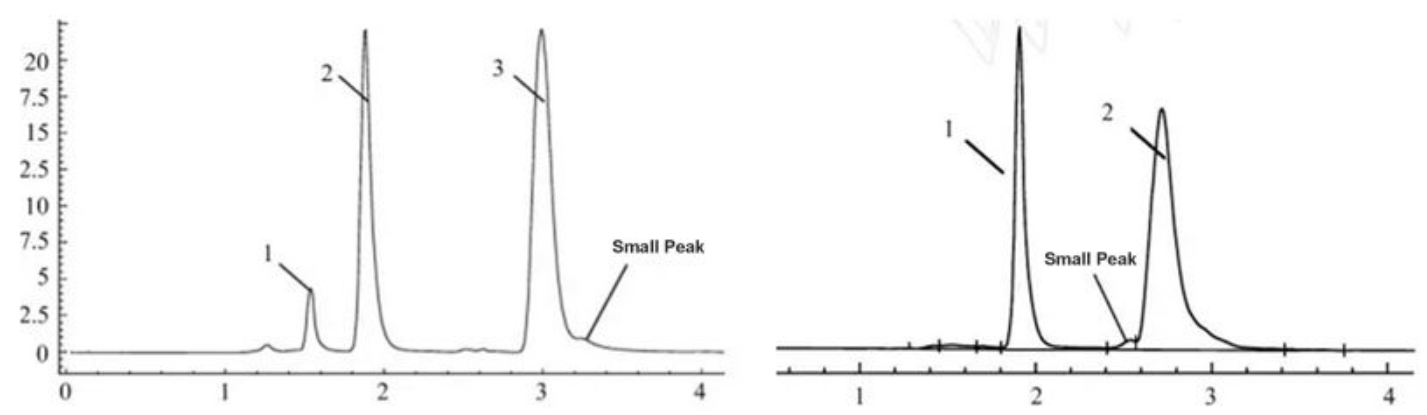
Potential Problems | Solution |
|---|---|
The sample is not pure | Using different mobile phase and HPLC columns to separate and compare samples, and select suitable separation conditions. |
Column collapse of analysis column or protective column | This is a common situation. Change the analysis column or guard column, then compare the peak shape. |
Column capacity decrease | After a long time of usage, there are some strong retention components adsorbed in the column, and small injection volumes tend to have peak splitting. The problem can be ameliorated by cleaning the column with a solvent with strong elution ability or by just replacing the column. |
The sample solvent does not match the mobile phase or the injection volume is too large | When the polarity of the sample solvent is larger than that of the mobile phase, sometimes the peak deformation and splitting phenomenon are easy to occur even if the sample volume is small. It is recommended to dissolve the sample with the mobile phase. |
Improper mobile phase | This situation is rare. Some samples under specific chromatographic conditions may have a dynamic equilibrium structure, and the double peak, the double peak can not be separated completely. Change the chromatographic conditions, especially the pH value to make the peak shape normal. |
Sample decomposition | Unstable samples will be changed into other substances during chromatographic separation and double peaks occur. So using the sample treatment methods or change chromatographic separation conditions. |
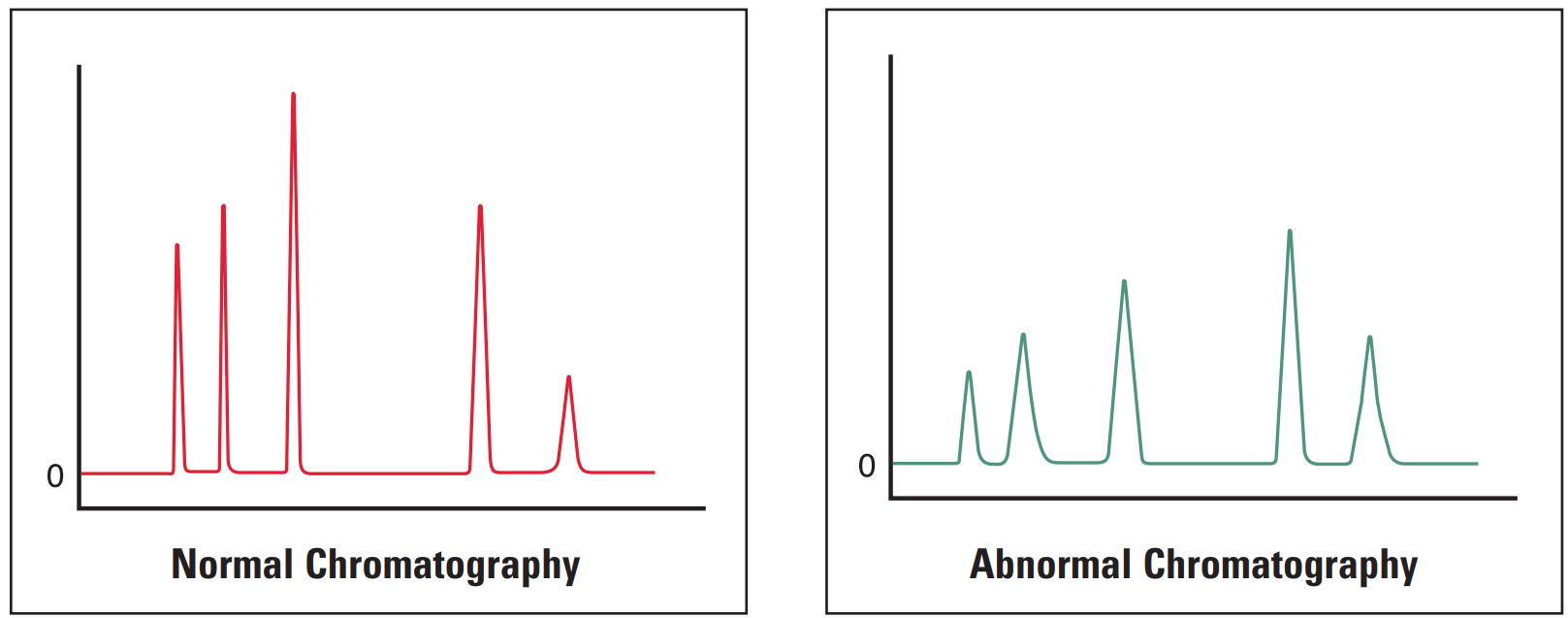
All Peaks Broad #
Thermo Guide #
Broad peaks (all) are most often due to errors in instrumentation or column. It is worthwhile investigating the column and guards first as they often are the critical part of the system.
Sigma-Aldrich Guide #
Problem | Probable Cause | Remedy/Comments |
|---|---|---|
|
1. Mobile phase composition changed. |
1. Prepare new mobile phase. |
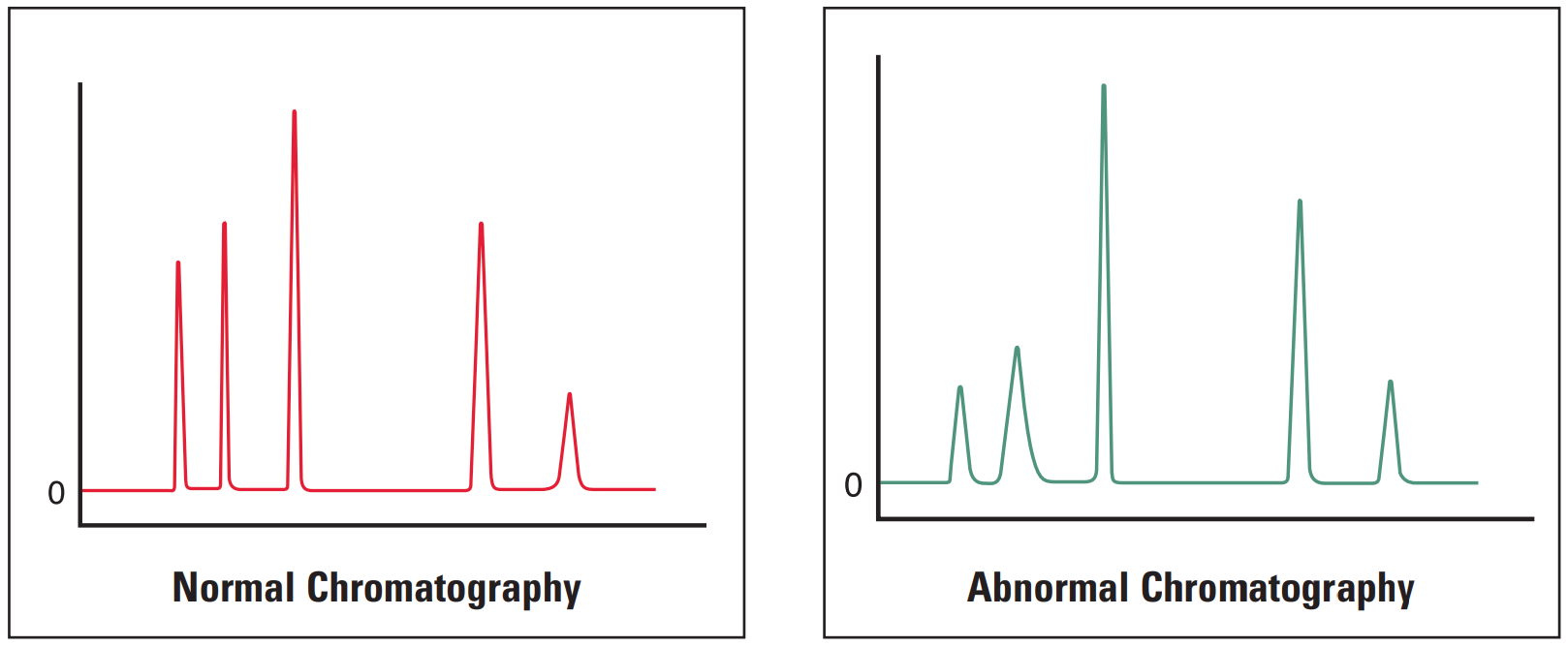
Early Eluting Peaks Broad #
Broad early eluting peaks are most commonly associated with sample overload or incorrect system plumbing.
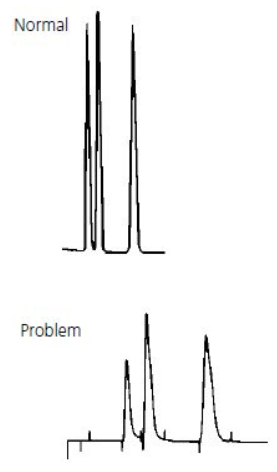
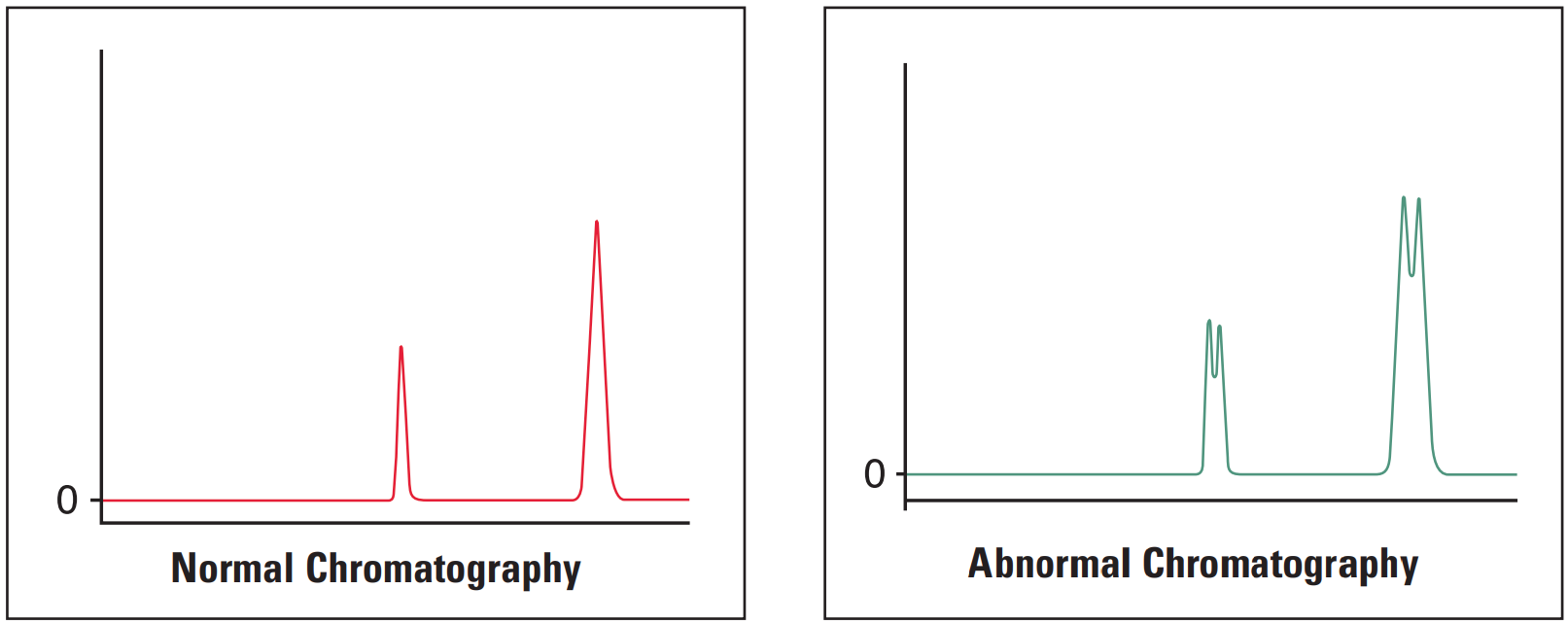
All Peaks Doubling #
Thermo Guide #
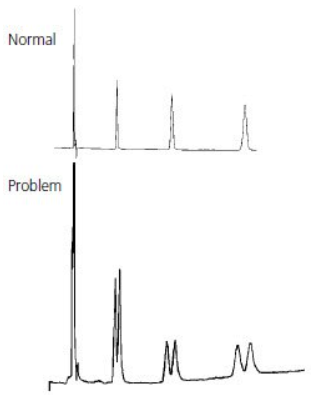
The most common cause of peak doubling can be either blockage prior to the column or column or guard voiding.
Galak #
- Column
If every peak has a double peak in the sample analysis process (the shorter the appearance time, the less likely it is to have a double peak), especially the analysis of the pure sample, there is a problem in the column (column head damaged, stationary phase in the column head dirty or lost).
If the sample injection volume is small and the column works well, the peak is a large peak with a small peak, it is not trailing. This may be caused by the column block. Wash the column with mobile phase solvent or acid cleaning or other solvents in a reverse direction, the block in the column will be washed away. Forward direction washing sometimes also can solve this problem.
If the peak shape is trailing with little difference between two peaks, it may be caused by the dirty or loss of the stationary phase in the column header.
- Solvent polarity and injection volume
For reversed-phase chromatography, the common mobile phases are methanol, acetonitrile, and water. Other additives are added to improve the separation performance. The sample is generally dissolved in a soluble solvent with the mobile phase, and the best solution is to use the mobile phase. When the solvent is strong polarity reagent (such as pure methanol, acetonitrile, pure ethanol) and the analysis system is mainly water, the double peaks phenomenon will occur with a large injection volume of pure sample (such as quantitative tube for 20ul): a peak combine with a smaller peak (different every time) with a tail, the retention time (relative to less sample quantity) will be earlier. If the injection volume is reduced by more than half, the peak shape will return to normal. This is because the solvent polarity of the sample is too different from that of the mobile phase, the mobile phase has no time to dilute it to reach equilibrium.
Another reason is that the injection volume is not necessarily large, but the absolute amount is large, and the two peaks on the chromatogram are close together, basically at the same height, without trailing (if the peak appearance time is very short, it may also be a column problem). If it is caused by a large injection and overload column, dilute the sample before injection.
- Sample characteristics
Some samples, due to the characteristics of their chemical structure, have the phenomenon of tautomerism, and the tautomerism can not be separated, but in a dynamic equilibrium existence. In the chromatographic analysis, under a specific condition, a substance will appear double peaks. These double peaks are very close with the same height and no tail. The double peak phenomenon will disappear when the condition is changed (especially pH).
- Device parameters
The parameters of the record are generally set internally and do not need to be modified without special needs. The HPLC recording time is generally 5ms. If the recording interval is shortened, one peak will become two or more peaks.
- Column contamination
- Frit block
- Column head collapsing (liquid phase pH≥7). Choose special columns will solve it.
- Solvent effect. The solvent is more polar than the mobile phase. It is also decided by the sample injection volume.
- Bubbles in the detector
Sigma-Aldrich Guide #
Problem | Probable Cause | Remedy/Comments |
|---|---|---|
|
1. Contamination on guard or analytical column inlet. |
1. Remove guard column (if present) and attempt analysis. Replace guard column if necessary. If analytical column is obstructed, reverse and flush. If problem persists, column may be clogged with strongly retained contaminants. Use appropriate restoration procedure (Table 2). If problem still persists, inlet frit is probably (partially) plugged. Change frit or replace column. |
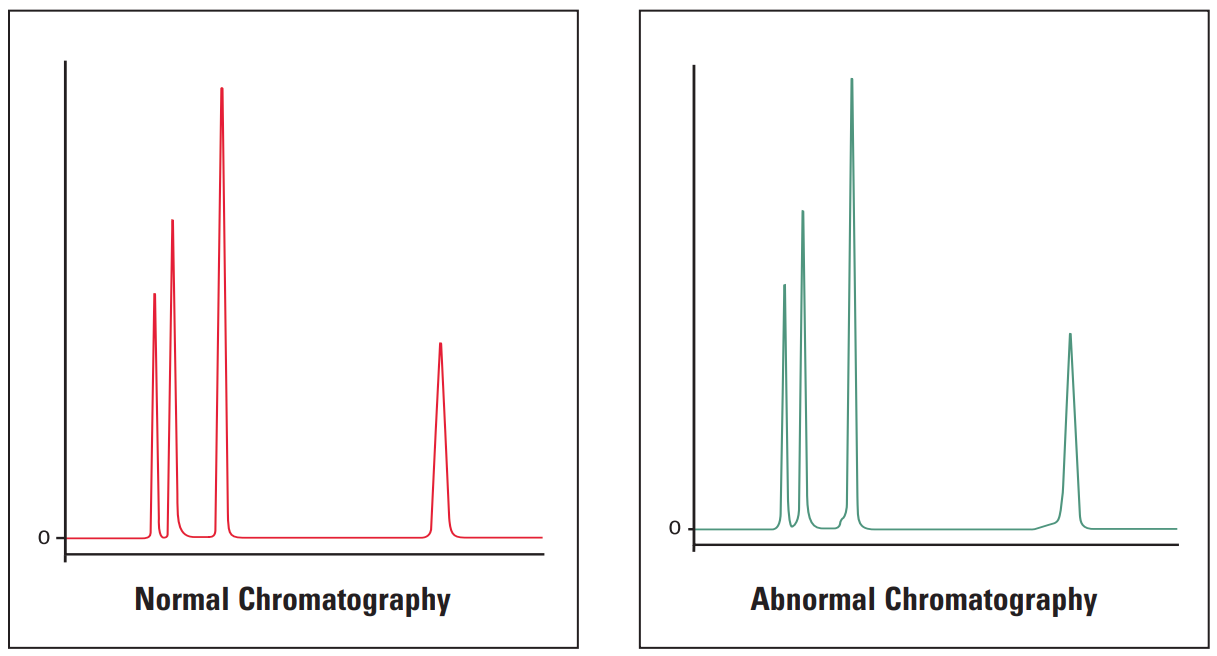
Fronting Peaks #
Thermo Guide #
Fronting peaks are very often due to large injection volumes of a sample that is dissolved in solvents that are incompatible with the mobile phase being used. The next most common cause of peak fronting is a voided or contaminated guard or column.
Galak Guide #
Potential Problem | Solution |
|---|---|
Improper solvent. | Choosing the appropriate solvent. |
The sample is overloaded. | Reduce the injection volume. |
The column temperature is too low. | Raising the column temperature. |
Column damaged. | Replace column. |
The main reason for the tailing peak and the leading peak is the improper selection of the mobile phase, which can be better improved by adjusting the polarity of the mobile phase or adding acid appropriately. Generally, acid and base in the mobile phase have a great influence on the tailing peak and the leading peak. The leading peak may be caused by column overload, and the tailing peak may be caused by sample contamination. Choosing the appropriate mobile phase and adjusting the pH value will improve this situation. Tailing peak is related to the column, maybe overload, dilute the sample, or use a new column. Sometimes the tailing peak is caused by the unseparated impurities with similar organic properties. You may need to optimize the analysis method or replace the column. Tailing peaks may also cause by column effect decline or column collapse after long column use time. Sometimes, tailing peaks are related to the properties of the sample, which some chemicals need to be added in the mobile phase to optimize the peak shape (depend on the specific situation).
Sigma-Aldrich Guide #
Problem | Probable Cause | Remedy/Comments |
|---|---|---|
|
1. Column overloaded. |
1. Inject smaller volume (e.g., 10 μL vs. 100 μL). Dilute the sample 1:10 or 1:100 fold in case of mass overload. |
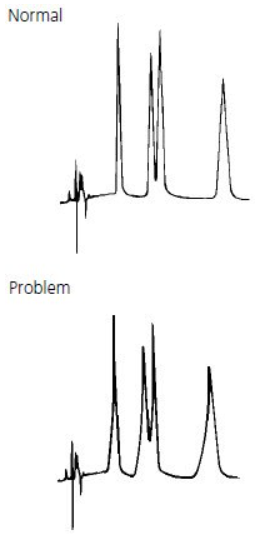
Tailing Peaks #
Thermo Guide #
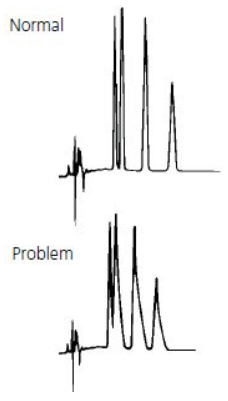
Tailing peaks are typically caused by column degradation or inlet contamination. Carefully maintained columns and guards will considerably reduce the incidence of tailing peaks.
Galak Guide #
Potential Problem | Remedy/Comments |
|---|---|
Interference peak. |
Separation under optimized conditions. |
Column collapse. |
Replace column. |
Mobile phase pH is not appropriate. |
Adjust the pH value. Add additives to eliminate secondary effects (interaction between components in the mobile phase and the column). |
Improperly connected pipeline. |
Large dead volume. Reconnect the pipeline. |
The injection volume is too large. |
Reduce it. |
The main reason for the tailing peak and the leading peak is the improper selection of the mobile phase, which can be better improved by adjusting the polarity of the mobile phase or adding acid appropriately. Generally, acid and base in the mobile phase have a great influence on the tailing peak and the leading peak. The leading peak may be caused by column overload, and the tailing peak may be caused by sample contamination. Choosing the appropriate mobile phase and adjusting the pH value will improve this situation. Tailing peak is related to the column, maybe overload, dilute the sample, or use a new column. Sometimes the tailing peak is caused by the unseparated impurities with similar organic properties. You may need to optimize the analysis method or replace the column. Tailing peaks may also cause by column effect decline or column collapse after long column use time. Sometimes, tailing peaks are related to the properties of the sample, which some chemicals need to be added in the mobile phase to optimize the peak shape (depend on the specific situation).
Sigma-Aldrich Guide #
Problem | Probable Cause | Remedy/Comments |
|---|---|---|
|
1. Sample reacting with active sites. |
1. First check column performance with standard column test mixture. If results for test mix are good, add ion pair reagent or competing base or acid modifier. |
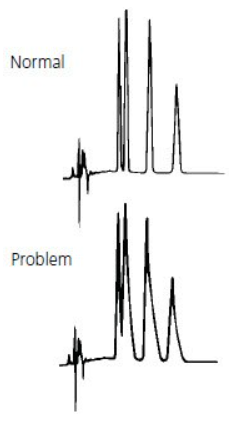
Problem | Probable Cause | Remedy/Comments |
|---|---|---|
|
1. Guard or analytical column contaminated/worn out. |
1. Remove guard column (if present) and attempt analysis. Replace guard column if necessary. If analytical column is source of problem, use appropriate restoration procedure (Table 2). If problem persists, replace column. |
Flat Topped Peaks #
Flat-topped peaks are most often caused by either large injection volumes of dilute sample or by small injection volumes of strong sample solution.

Change in Peak Height (one or more peaks) #
Problem | Probable Cause | Remedy/Comments |
|---|---|---|
|
1. One or more sample components deteriorated or column activity changed. |
1. Use fresh sample or standard to confirm sample as source of problem. If some or all peaks are still smaller than expected, replace column. If new column improves analysis, try to restore the old column, following appropriate procedure (Table 2). If performance does not improve, discard old column. |
Rounded Peaks #
Sigma-Aldrich Guide #
Problem | Probable Cause | Remedy/Comments |
|---|---|---|
|
1. Detector operating outside linear dynamic range. |
1. Reduce sample volume and/or concentration. |
Extra Peaks #
Thermo Guide #
Extra peaks in chromatograms are more often than not due to contamination or degradation of the sample, mobile phase or column. To check if the extra peak(s) is/are in the sample alone, perform a blank injection of sample solvent. The peak(s) should be absent.
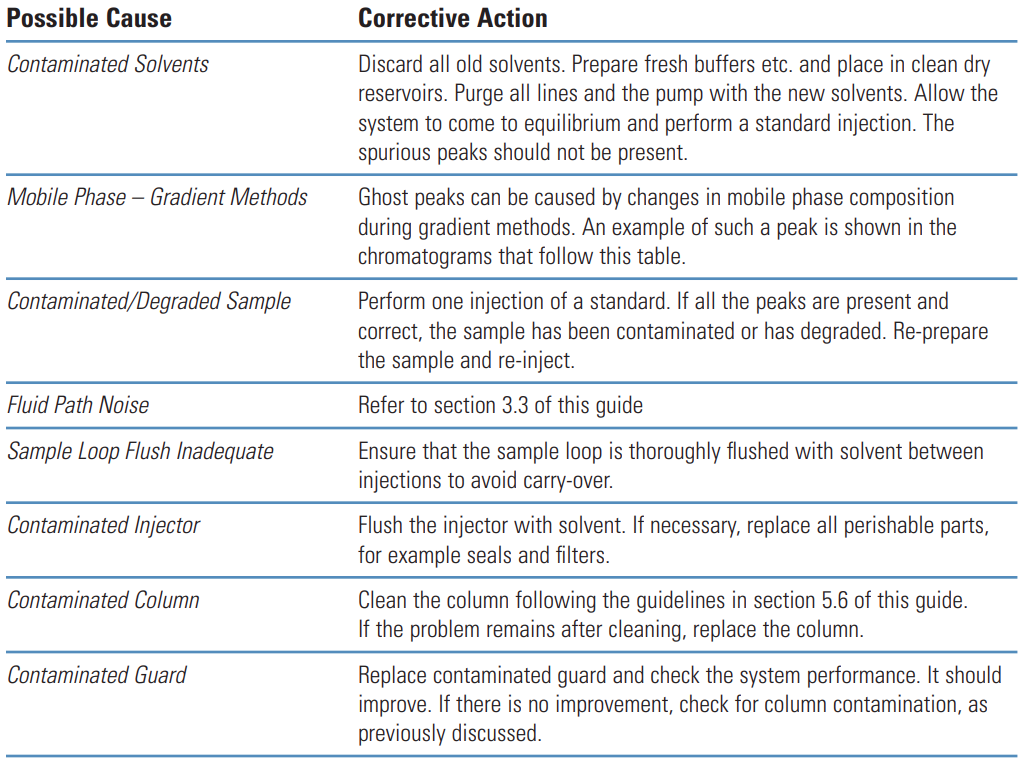
Negative Peaks #
Thermo Guide #
Negative peaks are most often caused by differences in refractive index between the sample solvent, sample and mobile phase.
They are also caused after routine maintenance when the system has not been reconfigured correctly.
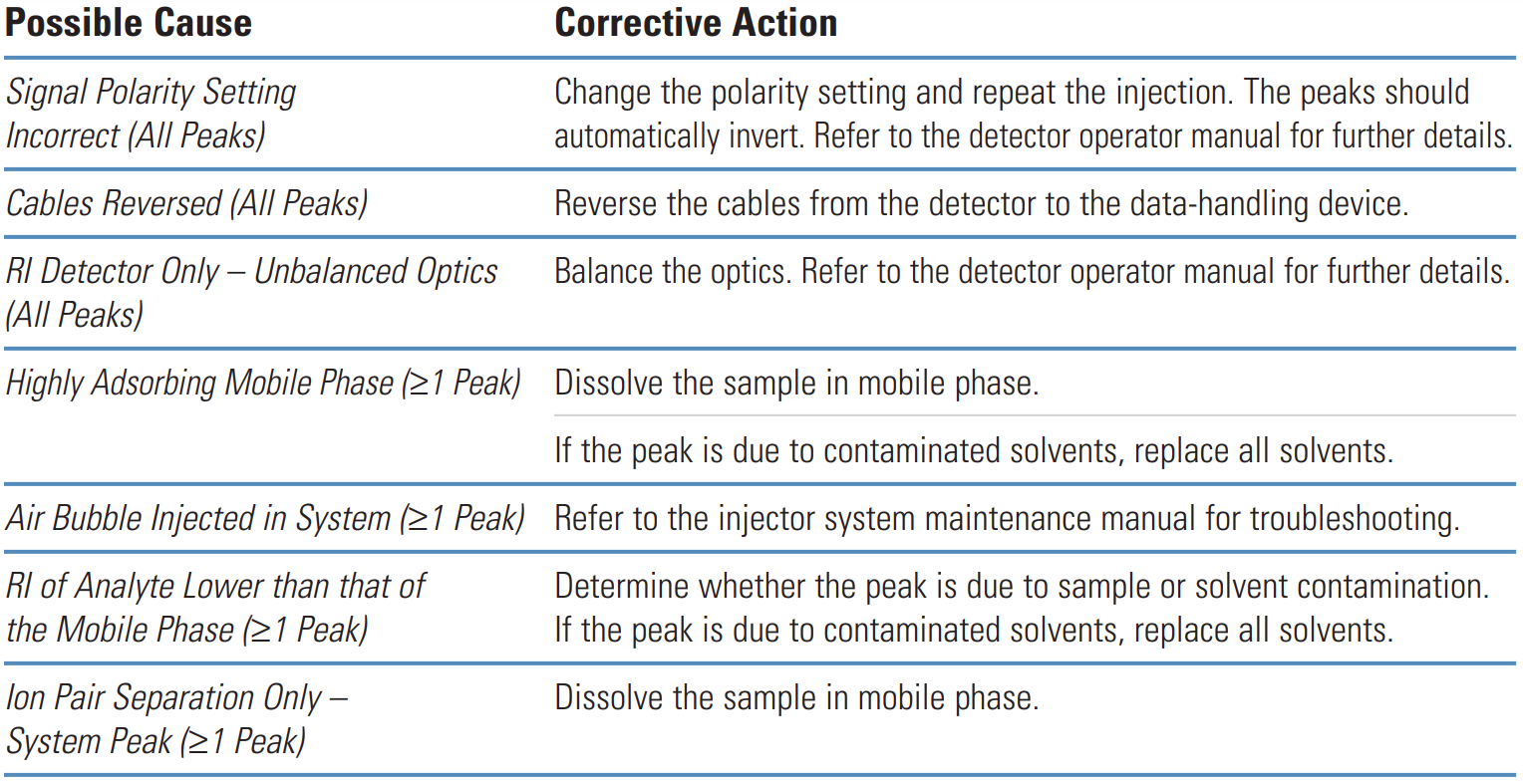
Galak Guide #
Potential Problem | Remedy/Comments |
|---|---|
The mobile phase absorption background value is too high. |
Change the detection wavelength appropriately. |
Air is entered during the injection. Using exhaust treatment. |
Re-inject when the baseline is stable. |
The absorption of sample components is lower than that of the mobile phase. |
Change the mobile phase or the detection wavelength. |
The solution of the sample preparation is different from the mobile phase. |
Re-prepare the sample with the same solvent as the flow, or dilute the sample. |
Sigma-Aldrich Guide #
Problem | Probable Cause | Remedy/Comments |
|---|---|---|
|
1. Recorder leads reversed. |
1. Check polarity. |
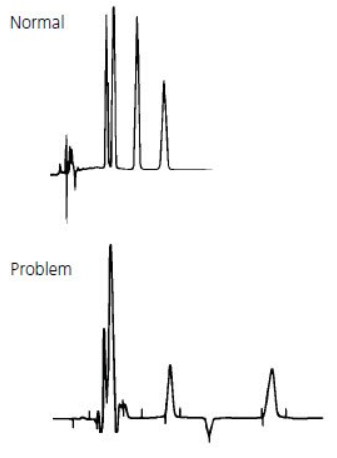
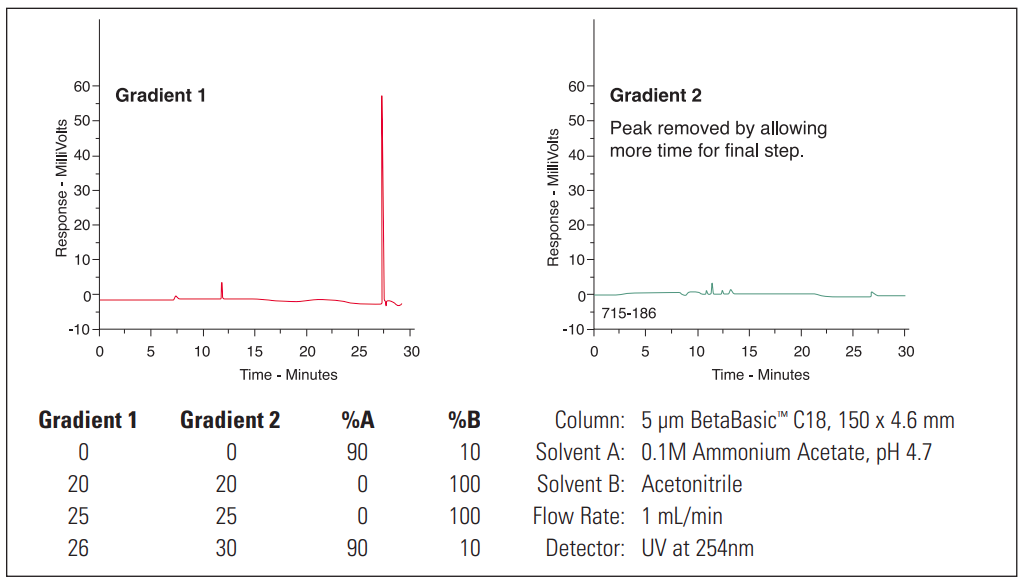
Ghost Peak #
Galak Guide #
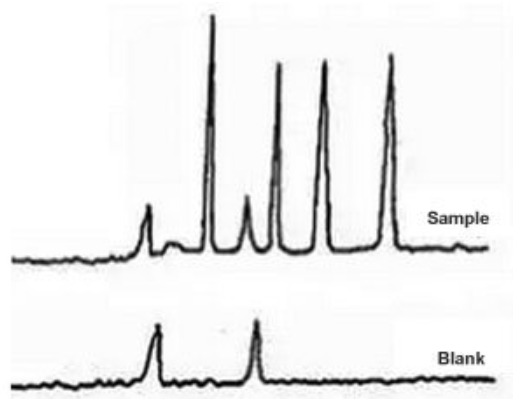
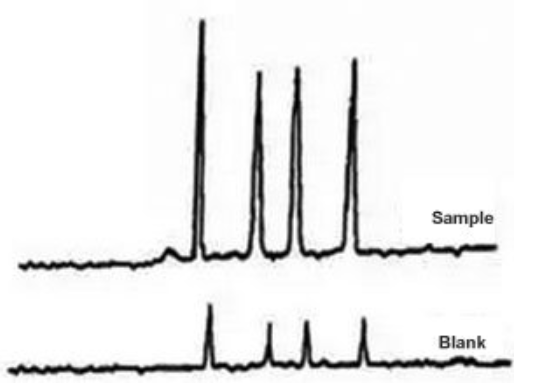
Sigma-Aldrich Guide #
Problem | Probable Cause | Remedy/Comments |
|---|---|---|
|
1. Contamination in injector or column. |
1. Flush injector between analyses (a good routine practice). If necessary, run strong solvent through column to remove late eluters. Include final wash step in gradient analyses, to remove strongly retained compounds. |
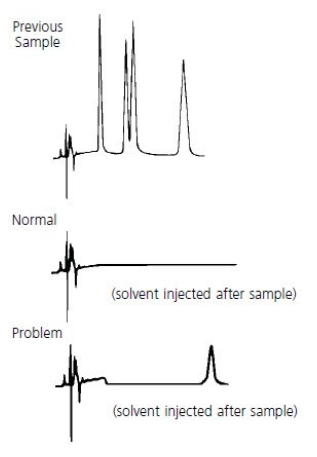
Diagrams for Troubleshooting Abnormal Peak Shape #
stateDiagram-v2 State1: The state with a note State1 --> State2
No Peak
Missing Peak]) --> 1.1[Sample] 1.1 --> 1.1.1[no sample?] 1.1 --> 1.1.2[sample deteriorated?] 1.1 --> 1.1.3[wrong sample?] 1.1 --> 1.1.4[Wrong position?] 1.1 --> 1.1.5[Insufficient sample?] 1.1 --> 1.1.6[Needle blocked?] A([Smaller peak
No Peak
Missing Peak]) --> 1.2[Mobile phase] 1.2 --> 1.2.1[Is solvent correct?] 1.2 --> 1.2.2[correct proportions of solvents?] 1.2 --> 1.2.3[correct gradient or isocratic path] --> 1.2.3.1[gradient sufficient?] 1.2 --> 1.2.4[no flow? low flow?] 1.2.4 --> 1.2.4.1[flow rate accurate?] 1.2.4 --> 1.2.4.2[pump works?] 1.2.4 --> 1.2.4.3[blockage?] --> 1.2.4.2.1[pressure?] A([Smaller peak
No Peak
Missing Peak]) --> 1.3[Column] 1.3 --> 1.3.1[Resolution lost?] --> 1.3.1.1[check the efficiency of the original column] 1.3 --> 1.3.2[column dimension? particle size?]