Transcripts #
Slide #
So let’s in somewhat the same manner, talk about peak shape. So we’ll talk about where why we get abnormal peak shapes. And then in the same way, some cases. So this is what I call the weirdest peak contest. And and leading up to this grace and Mike, right, and I exchanged some examples, and I still like mine at best. In case, you, you didn’t see it. That’s the problem right there.
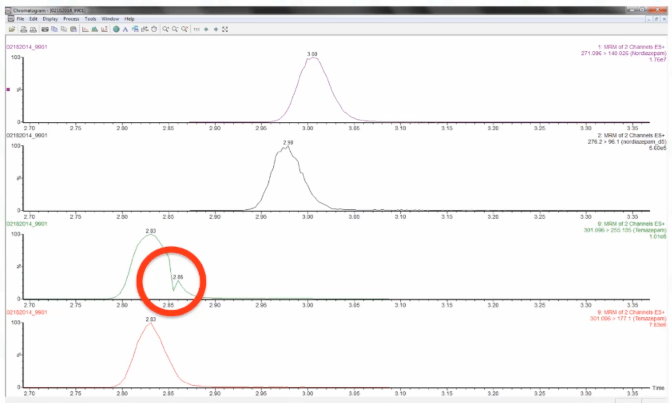
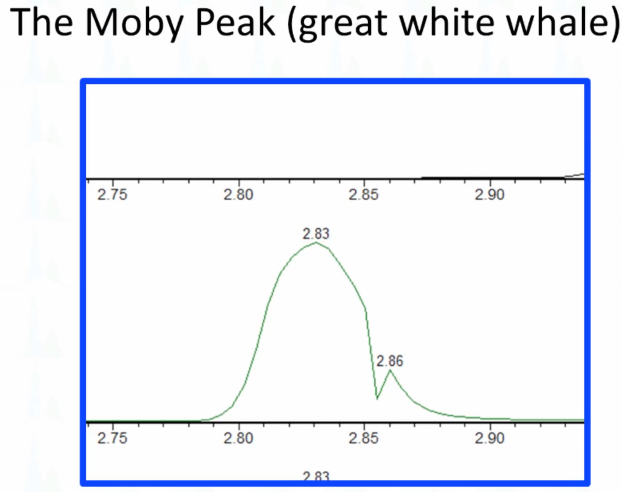 And if you come from the US, you know that everybody has to read at some point, when you're going to school, a Herman Melville's Moby Dick, and it's about a whale. And I think this looks like a whale story. So I call this the Mobi peak, the great white whale. And we see this on our Waters TQS system, very reproducible, sometimes for a whole batch where we've had a slight shift in retention time. And this lines up perfectly with where there is an end of a function, or what is called a period with AB Sciex. And so what it seems like to us is that when there's especially when there's a lot of functions lining up or starting, and it happens at a certain part of the peak, you'll have a signal drop out, it's like there's a delay in passing of the signal from the mass spec to the LC. If you smooth your data, you will never see it. if you don't smooth, you'll see it now. And then the current version that we're using SCN 905 MassLynx isn't supposed to have that. But it does create some very interesting peaks.
And if you come from the US, you know that everybody has to read at some point, when you're going to school, a Herman Melville's Moby Dick, and it's about a whale. And I think this looks like a whale story. So I call this the Mobi peak, the great white whale. And we see this on our Waters TQS system, very reproducible, sometimes for a whole batch where we've had a slight shift in retention time. And this lines up perfectly with where there is an end of a function, or what is called a period with AB Sciex. And so what it seems like to us is that when there's especially when there's a lot of functions lining up or starting, and it happens at a certain part of the peak, you'll have a signal drop out, it's like there's a delay in passing of the signal from the mass spec to the LC. If you smooth your data, you will never see it. if you don't smooth, you'll see it now. And then the current version that we're using SCN 905 MassLynx isn't supposed to have that. But it does create some very interesting peaks.
Slide: Narrow Peaks = Higher Efficiency #
Okay, so we always want narrower peaks, we say that’s higher efficiency. It is the chromatographic term used to characterize peak width, you’ll hear people talk about it either in units of time, or in units of mobile phase sometimes. And it’s pretty simple. Narrower peaks are better than wide peaks. it’s that simple, better signal to noise, lower quantitation limits. So this would be if you had the same amount of analyte, depending on things, how this is the same amount. But here it’s spread out. And I think you can, it’s kind of obvious that the skinnier peak is going to give you a better detection limits about the only time you can have it too narrow if your mass spec can’t scan fast enough. And we’ll talk about what that might look like. And also, of course, it’s going to give you the potential for better resolution. So this is the situation where it’s clearly not acceptable for quantitation. In here, it’s baseline resolved, we didn’t have any difference and selectivity, both pigs are still coming out at exactly the same retention time. But we’ve had an improvement in efficiency. And so it gives us resolution.
Why abnormal peaks? #
 So of the possible things that can cause an abnormal peak, in my experience, it's very heavily coming from the LC very just occasionally, is it going to be ms related? And I've kind of called it a sample prep faux pas, because if you did your method development work acceptably, then it shouldn't happen from sample prep unless somebody makes a mistake.
So of the possible things that can cause an abnormal peak, in my experience, it's very heavily coming from the LC very just occasionally, is it going to be ms related? And I've kind of called it a sample prep faux pas, because if you did your method development work acceptably, then it shouldn't happen from sample prep unless somebody makes a mistake.
Dwell Time Issue #
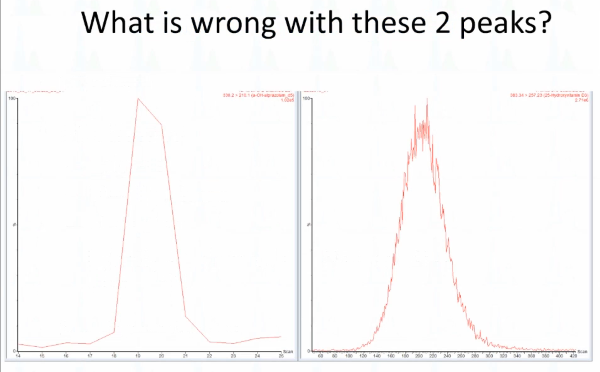
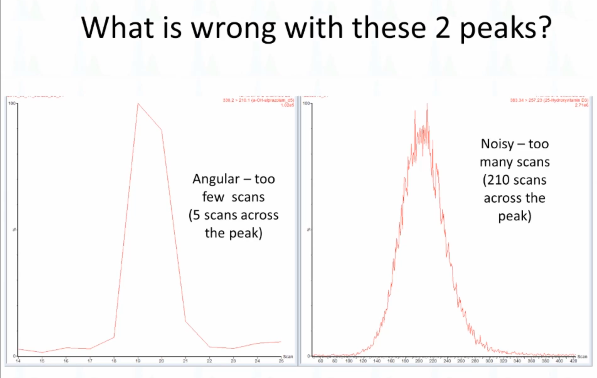 Okay, so what is wrong with these two peaks? How about this one on the left? Do you think that looks like a normal peak? What don't you like about that peak? What's wrong? Right, so it's very `angular looks geometric`. Right? How about this one over here? Anyway, anybody ever seen that before? Took me a long time to finally realize what it is? This is too few scans that's five scans across the peak? This is too many that's 210 scans across the peak? And maybe we'll you would know, my assumption is that essentially, you just have a lot of noise. I mean, so many things are arriving at that time that there's variance, but signal averaging is not good enough over that shorter. Yeah, yeah. So this is like if you have a two millisecond dwell time or something like that, and it kind of fat peak. Of course, I, these were generated mostly by my mistakes, that's where they get all right. So this is the scenario that we're talking about, which is that our dwell time is set too long. And so what I want everybody to remember is that the mass spec, it has a short attention span, if you like, so it looks here. And then for the rest of this time, it's looking at the other eight, nine or MRMS in this method. So here was MRM one, and then because it's looking elsewhere, it doesn't see what's happening until it gets back and looks again and by this time, we've already gotten almost to the apex of the peak. Okay, so that's what's happened when your dwell time is not right. And this problem, I would say, is much more typical and much more of a problem than this.
Okay, so what is wrong with these two peaks? How about this one on the left? Do you think that looks like a normal peak? What don't you like about that peak? What's wrong? Right, so it's very `angular looks geometric`. Right? How about this one over here? Anyway, anybody ever seen that before? Took me a long time to finally realize what it is? This is too few scans that's five scans across the peak? This is too many that's 210 scans across the peak? And maybe we'll you would know, my assumption is that essentially, you just have a lot of noise. I mean, so many things are arriving at that time that there's variance, but signal averaging is not good enough over that shorter. Yeah, yeah. So this is like if you have a two millisecond dwell time or something like that, and it kind of fat peak. Of course, I, these were generated mostly by my mistakes, that's where they get all right. So this is the scenario that we're talking about, which is that our dwell time is set too long. And so what I want everybody to remember is that the mass spec, it has a short attention span, if you like, so it looks here. And then for the rest of this time, it's looking at the other eight, nine or MRMS in this method. So here was MRM one, and then because it's looking elsewhere, it doesn't see what's happening until it gets back and looks again and by this time, we've already gotten almost to the apex of the peak. Okay, so that's what's happened when your dwell time is not right. And this problem, I would say, is much more typical and much more of a problem than this.
Inter-scan Delay #
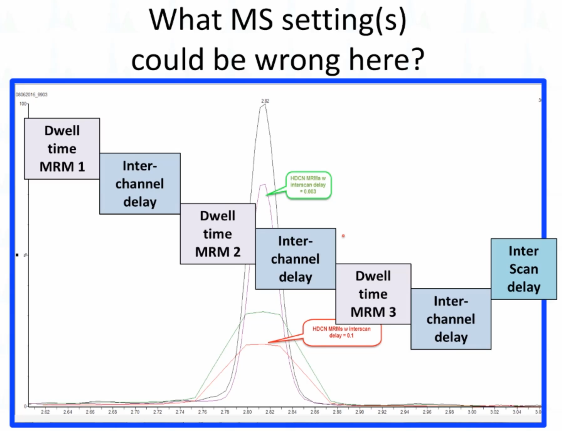 Okay, so I'm going to just tell you right up front, this is not a dwell time problem, nobody changed the dwell time problem here. But that peak looks kind of like the dwell time is too long, right? So what do you think could be the difference here, if you can read the fine print, it's too cheap. It tells you what's there. This is the same vial injected when the problem was discovered, and then after the fix. So when you're we always talk about duty cycle, right? I always felt like when I was first doing mass spec, if I could say duty cycle, it sounds kind of cool. You know, like I knew what I was talking about. But I didn't necessarily really know what I was talking about. And here's the issue, when we build a scan for a mass spec, we don't just have dwell time, depending on your vendor, they might call it a little something. But in between the two MRM, there's something where it's flushing out the collision cell, right, it's getting all the product ions from the previous scan out of the collision cell, so that for the next MRM, you don't have any what we would call crosstalk you don't have anything left for the last scan. So on a Waters TQS system, they call that an inter-channel delay, then we often will build a set of MRMs as a function as a period, whatever it is, you're grouping them together, right for what your purpose. And then at the end, there's something called the Interscan delay, typically, or at least there is with a Waters system. So what happened here is that we our service guy was on site, and he was troubleshooting something. And he thought maybe the problem was the Interscan delay, and he changed it from 3 msec. to 100 msec. And so that's what happens when you do that.
Okay, so I'm going to just tell you right up front, this is not a dwell time problem, nobody changed the dwell time problem here. But that peak looks kind of like the dwell time is too long, right? So what do you think could be the difference here, if you can read the fine print, it's too cheap. It tells you what's there. This is the same vial injected when the problem was discovered, and then after the fix. So when you're we always talk about duty cycle, right? I always felt like when I was first doing mass spec, if I could say duty cycle, it sounds kind of cool. You know, like I knew what I was talking about. But I didn't necessarily really know what I was talking about. And here's the issue, when we build a scan for a mass spec, we don't just have dwell time, depending on your vendor, they might call it a little something. But in between the two MRM, there's something where it's flushing out the collision cell, right, it's getting all the product ions from the previous scan out of the collision cell, so that for the next MRM, you don't have any what we would call crosstalk you don't have anything left for the last scan. So on a Waters TQS system, they call that an inter-channel delay, then we often will build a set of MRMs as a function as a period, whatever it is, you're grouping them together, right for what your purpose. And then at the end, there's something called the Interscan delay, typically, or at least there is with a Waters system. So what happened here is that we our service guy was on site, and he was troubleshooting something. And he thought maybe the problem was the Interscan delay, and he changed it from 3 msec. to 100 msec. And so that's what happens when you do that.
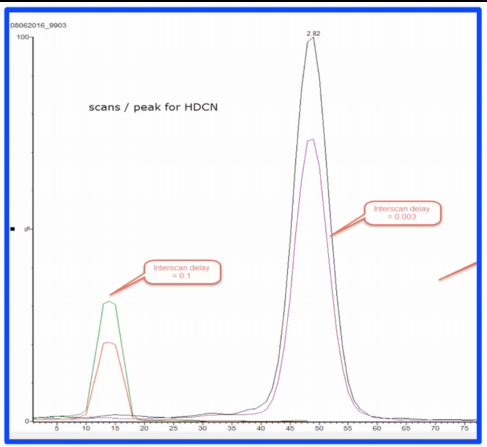 And if you look at this problem, now this axis is scan number rather than time, okay. And that's where it really stands out when I first pulled this up, and I forgot that I had left it in scan number. I'm like, why? that looks like they're different retention times know, what this is showing you is that for this particular period of time, the mass spec was scanning. And here when the when the Interscan delay was set too long. That's that flat top. So it's only eight, about eight scans across the page. Okay. Here where it normally is. It's now from about 40, to about 60. There's about what 28 or something or 18 across the peak. So just want to point out that it's not all about dwell time, this is a pretty unusual problem, but it can happen. The interesting thing is with this method, the early eluting peaks in our method were completely resolved. And so they only had a few scans and interesting, sorry, a few MRMs in the method. And so those peaks look normal. It wasn't until we got to a place, we had a bunch of overlapping ones. And so there was a lot of Interscan thrown into Interscan delay that this problem happened. We were lucky because I had seen it happen before otherwise, I would have been faked out by what? Why? How did that happen?
And if you look at this problem, now this axis is scan number rather than time, okay. And that's where it really stands out when I first pulled this up, and I forgot that I had left it in scan number. I'm like, why? that looks like they're different retention times know, what this is showing you is that for this particular period of time, the mass spec was scanning. And here when the when the Interscan delay was set too long. That's that flat top. So it's only eight, about eight scans across the page. Okay. Here where it normally is. It's now from about 40, to about 60. There's about what 28 or something or 18 across the peak. So just want to point out that it's not all about dwell time, this is a pretty unusual problem, but it can happen. The interesting thing is with this method, the early eluting peaks in our method were completely resolved. And so they only had a few scans and interesting, sorry, a few MRMs in the method. And so those peaks look normal. It wasn't until we got to a place, we had a bunch of overlapping ones. And so there was a lot of Interscan thrown into Interscan delay that this problem happened. We were lucky because I had seen it happen before otherwise, I would have been faked out by what? Why? How did that happen?
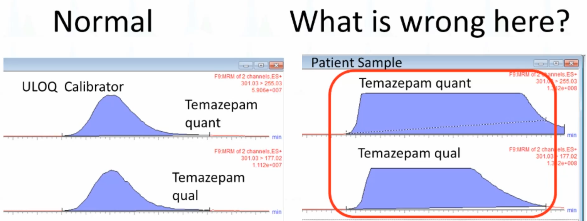 Okay. So this is a normal. Normally what peak look like for one of our analytes and a calibrator, Temazepam. if you're not used to the presentation of data with Waters, this is the quantifier, the qualifier and then the IS, the internal standard in peak review. So, What's wrong here? What's going on there? This is an obvious one, come on. Why does it look like the, you know, a high rise building? Yep. So the peak is saturated, right? So this is a really classic appearance for those of you who use Waters, you know that this 1.3 to eight as the limitation of the amount of signal. Okay, so that's pretty obvious. There's too much to me. Temazepam, somebody probably gave this person IV Valium.
Okay. So this is a normal. Normally what peak look like for one of our analytes and a calibrator, Temazepam. if you're not used to the presentation of data with Waters, this is the quantifier, the qualifier and then the IS, the internal standard in peak review. So, What's wrong here? What's going on there? This is an obvious one, come on. Why does it look like the, you know, a high rise building? Yep. So the peak is saturated, right? So this is a really classic appearance for those of you who use Waters, you know that this 1.3 to eight as the limitation of the amount of signal. Okay, so that's pretty obvious. There's too much to me. Temazepam, somebody probably gave this person IV Valium.
Ion Suppression or Matrix Effect #
 What's this? Why does this look like somebody took a bite out of it? So the retention time is the key. What do we know about ESI Ionization? Is there enough charge to go around all the time? No, there's not enough charge to go around. Right? We say that we have ion suppression. And so here what's happening is the Temazepam is causing `ion suppression` at the internal standard. Does that make sense to everybody? And it's for a very limited part of time, it's well this is eluding. So it starts out normal and then the Temazepam comes up and the internal standards kind of, oh, I'm lost. I don't have enough. I don't get enough charge. And then as soon as this goes out, then it starts to be normal again. We see this a lot with methamphetamine too, because methamphetamine comes at such high concentrations. So this is obviously something that you need to dilute or Before you analyze it again. But does that make sense to everybody, when you see this odd peak shape, we always think of `ion suppression` is coming from the matrix. But you can also get ion suppression from another analyte. In the method, sometimes we would see this odd shape, but you can't see any matching peak that's coming off. What would that say to you if you saw a shape like this, but there was nothing out saturated that was around what might be going on. When you design an MRM method, are you looking at every single thing that's in your sample? No, you're not. So what do you think might be going on? If you see that kind of a picture? So what? could `the matrix effect` that's what we that's what we would often say is that there's probably some other large drug like an antibiotic or something in there, that's coming off at that time, we can't see it. But it's still stealing charge away from our peak that we want to measure, right.
What's this? Why does this look like somebody took a bite out of it? So the retention time is the key. What do we know about ESI Ionization? Is there enough charge to go around all the time? No, there's not enough charge to go around. Right? We say that we have ion suppression. And so here what's happening is the Temazepam is causing `ion suppression` at the internal standard. Does that make sense to everybody? And it's for a very limited part of time, it's well this is eluding. So it starts out normal and then the Temazepam comes up and the internal standards kind of, oh, I'm lost. I don't have enough. I don't get enough charge. And then as soon as this goes out, then it starts to be normal again. We see this a lot with methamphetamine too, because methamphetamine comes at such high concentrations. So this is obviously something that you need to dilute or Before you analyze it again. But does that make sense to everybody, when you see this odd peak shape, we always think of `ion suppression` is coming from the matrix. But you can also get ion suppression from another analyte. In the method, sometimes we would see this odd shape, but you can't see any matching peak that's coming off. What would that say to you if you saw a shape like this, but there was nothing out saturated that was around what might be going on. When you design an MRM method, are you looking at every single thing that's in your sample? No, you're not. So what do you think might be going on? If you see that kind of a picture? So what? could `the matrix effect` that's what we that's what we would often say is that there's probably some other large drug like an antibiotic or something in there, that's coming off at that time, we can't see it. But it's still stealing charge away from our peak that we want to measure, right.
MS related #
Okay, so I would say for mass spec, what you want to do is keep in mind that quadrupole acquisition, MRM acquisition is discontinuous. Always keep that we think we look at smooth peaks. And it’s easy to forget that, that the MS is only looking for each analyte part of the time. So there’s only so much time to go around in the duty cycle. And then also when it comes to ionization, it’s kind of a dog eat dog. I’m not sure that it’s necessarily scientifically correct to say that there’s a limited amount of charge, and there’s competition. But that works for me to think about what’s going on with ion suppression.
Slide #
 Okay, so this is normal. And now we see that. what do you think's going on here?
If you've never had this happen in your lab, I would be amazed. So I'm going to tell you this is this is when `the guard column` is new. And this is when `the guard column` has 700 injections going on. So what? Yes, exactly. Thank you. So this is an aged column or guard column will look like that. And we first see the peak widen and then it starts to split like that. Aged guard column. I primed here to say that right. And so if you reinjected after you put on a new guard, if you do it the right way, then it looks good again.
Okay, so this is normal. And now we see that. what do you think's going on here?
If you've never had this happen in your lab, I would be amazed. So I'm going to tell you this is this is when `the guard column` is new. And this is when `the guard column` has 700 injections going on. So what? Yes, exactly. Thank you. So this is an aged column or guard column will look like that. And we first see the peak widen and then it starts to split like that. Aged guard column. I primed here to say that right. And so if you reinjected after you put on a new guard, if you do it the right way, then it looks good again.
Slide #
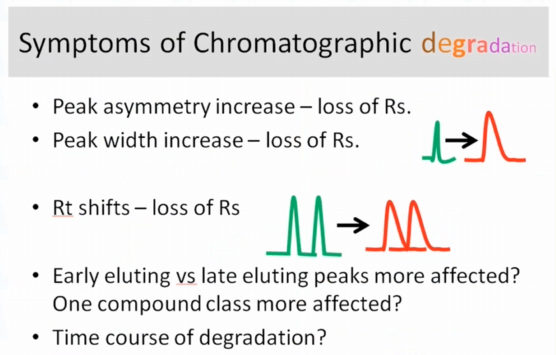 Okay, so if we're talking about symptoms, we've now switched, obviously, to LC problems, we're talking about symptoms of a degradation. One of the things that I like I like Russ Graham, all it says in his course, is the column is a consumable. And so we might think, I can't throw it away. And it has to be said, I have to make it last for 10,000 injections, we have to balance that with how it's affecting the rest of our assay. So we if you have a peak asymmetry increase, if you have a peak increase with the loss of resolution that's going to happen when you have aging of your consumables, same thing, I would say consent to retention time shift. And I've actually seen for us often we'll see things come off a little sooner, the retention time is a little shorter as the column or `the guard column` ages, presumably, to me, because at the head of the column, you have all this matrix residue deposited. I've also seen this scenario where an early eluding peak might be affected, and other peaks in the chromatogram might not be affected. And that can be a little confusing, right, because you wouldn't think it would affect the whole run. But it kind of makes sense, because the early eluting stuff doesn't necessarily comes up sooner to the con because it's not retained as well, right. So it kind of makes sense that it would be that one might be affected first. And the other thing I think that's useful to know is look at the time course of the degradation.
Okay, so if we're talking about symptoms, we've now switched, obviously, to LC problems, we're talking about symptoms of a degradation. One of the things that I like I like Russ Graham, all it says in his course, is the column is a consumable. And so we might think, I can't throw it away. And it has to be said, I have to make it last for 10,000 injections, we have to balance that with how it's affecting the rest of our assay. So we if you have a peak asymmetry increase, if you have a peak increase with the loss of resolution that's going to happen when you have aging of your consumables, same thing, I would say consent to retention time shift. And I've actually seen for us often we'll see things come off a little sooner, the retention time is a little shorter as the column or `the guard column` ages, presumably, to me, because at the head of the column, you have all this matrix residue deposited. I've also seen this scenario where an early eluding peak might be affected, and other peaks in the chromatogram might not be affected. And that can be a little confusing, right, because you wouldn't think it would affect the whole run. But it kind of makes sense, because the early eluting stuff doesn't necessarily comes up sooner to the con because it's not retained as well, right. So it kind of makes sense that it would be that one might be affected first. And the other thing I think that's useful to know is look at the time course of the degradation.
Frit or column aging #

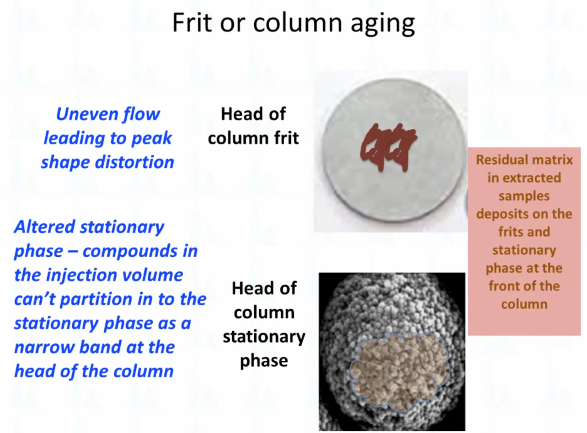 So this is how I think of it is to what's going on, which is you're getting some matrix residue deposited on the frit. And this is an electron micrograph for the silica particles at the head of the column. And so you have both uneven flow and uneven partitioning into that stationary phase instead of that tight, narrow band that you want in order to get a good peek.
So this is how I think of it is to what's going on, which is you're getting some matrix residue deposited on the frit. And this is an electron micrograph for the silica particles at the head of the column. And so you have both uneven flow and uneven partitioning into that stationary phase instead of that tight, narrow band that you want in order to get a good peek.
Slide #
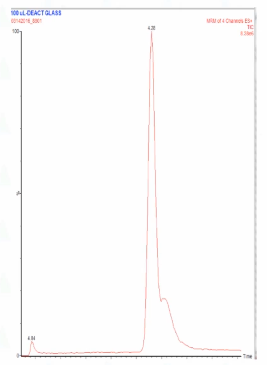 So this is a testosterone assay that I was working on. And I had fixed one problem and I thought everything was great. And then I came back from coffee and I saw this. so what do you think what could be going on? We already did column degradation. So you know it's not that right as to be another choice. What might go on? I will tell you that I did touch the system. I did something to the system before I went for coffee. So what might have done that caused the peak to look like that?
Yes, Actually, somebody did something with connectors, it wasn't me to start with.
So this is a testosterone assay that I was working on. And I had fixed one problem and I thought everything was great. And then I came back from coffee and I saw this. so what do you think what could be going on? We already did column degradation. So you know it's not that right as to be another choice. What might go on? I will tell you that I did touch the system. I did something to the system before I went for coffee. So what might have done that caused the peak to look like that?
Yes, Actually, somebody did something with connectors, it wasn't me to start with.
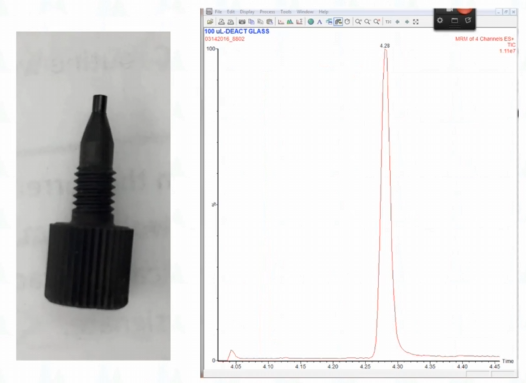 I intervened. And so I ended up having to put a new fitting on there. And then the peak looked like this. So this was a long story of several people did things with plumbing. And one fitting was replaced. And as we talked about, there was probably a gap here. And so that's what happened with the peak, typically the peak is about three seconds wide. That was interestingly enough the outlet. I know I thought that was pretty weird. Actually, here's the truth of the matter if you've ever worked with the Waters, I don't know what kind of Waters column manager we have. But it has this little short tubing. And it has this fitting that doesn't even have a wrench. How do you Yeah, and so if you're me, when you're trying to hold that, you're always turning the column manager off by mistake, and then you have to reset communications. So it's really terrible to do. And the other fitting that's hard to do is it goes into a post column valve. And essentially, you have to have a little teeny fingers to work there. So I changed the the post column connection because it was easy. And that turned out to be what was wrong, but most of the time it isn't there.
I intervened. And so I ended up having to put a new fitting on there. And then the peak looked like this. So this was a long story of several people did things with plumbing. And one fitting was replaced. And as we talked about, there was probably a gap here. And so that's what happened with the peak, typically the peak is about three seconds wide. That was interestingly enough the outlet. I know I thought that was pretty weird. Actually, here's the truth of the matter if you've ever worked with the Waters, I don't know what kind of Waters column manager we have. But it has this little short tubing. And it has this fitting that doesn't even have a wrench. How do you Yeah, and so if you're me, when you're trying to hold that, you're always turning the column manager off by mistake, and then you have to reset communications. So it's really terrible to do. And the other fitting that's hard to do is it goes into a post column valve. And essentially, you have to have a little teeny fingers to work there. So I changed the the post column connection because it was easy. And that turned out to be what was wrong, but most of the time it isn't there.
Slide #
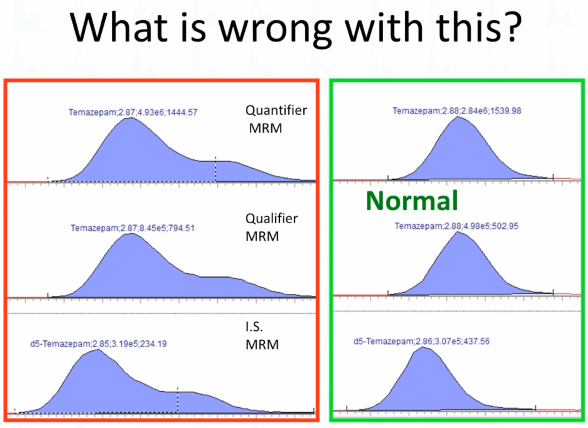
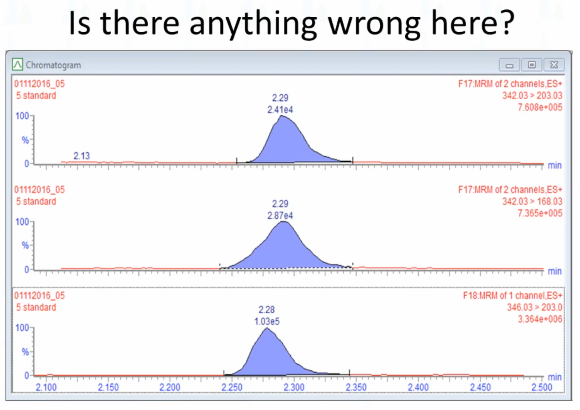 Okay, what do you think is wrong with this picture? There is something wrong with it. Let me make it easy for you. Do you think this one's okay. That peak good? How many hands hands? How many people think that's a good peek? Okay. All right. How about this bottom one? The internal standard? You think the internal standard picks? Okay. Okay, so kind of made it easy. So it must be this lag, right? What don't you like about this peak? Kind of spread out? Okay, so it looks like it maybe has a shoulder on it? Would you think if if if there was something wrong with the plumbing or something along like that, would you expect it to affect just one peak rather than all three? I would expect it to. So this seems like something other than bad plumbing, I would say.
Okay, what do you think is wrong with this picture? There is something wrong with it. Let me make it easy for you. Do you think this one's okay. That peak good? How many hands hands? How many people think that's a good peek? Okay. All right. How about this bottom one? The internal standard? You think the internal standard picks? Okay. Okay, so kind of made it easy. So it must be this lag, right? What don't you like about this peak? Kind of spread out? Okay, so it looks like it maybe has a shoulder on it? Would you think if if if there was something wrong with the plumbing or something along like that, would you expect it to affect just one peak rather than all three? I would expect it to. So this seems like something other than bad plumbing, I would say.
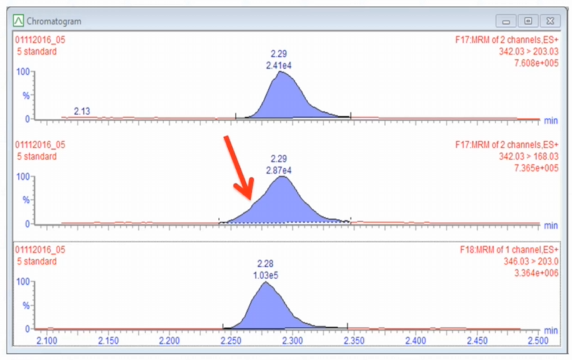 Right there. That's the problem. So if you inject the blank, or if we look at the blank that was injected with this batch, there should be no peak here. So right away, we know there's something wrong, right?
Right there. That's the problem. So if you inject the blank, or if we look at the blank that was injected with this batch, there should be no peak here. So right away, we know there's something wrong, right?
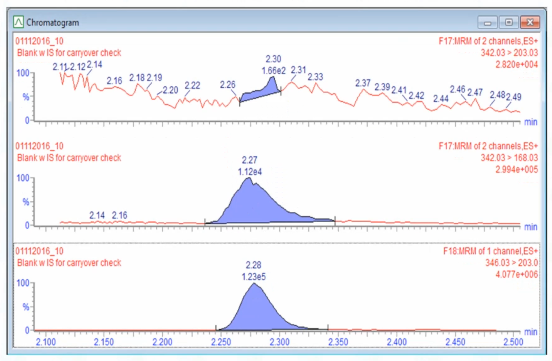 There's an interfering peak. And so when we first saw this, I got worried that maybe something had happened with the system. And then I just did what we'll talk about, which is I looked at the last batch, and I looked at the last set of batches.
There's an interfering peak. And so when we first saw this, I got worried that maybe something had happened with the system. And then I just did what we'll talk about, which is I looked at the last batch, and I looked at the last set of batches.
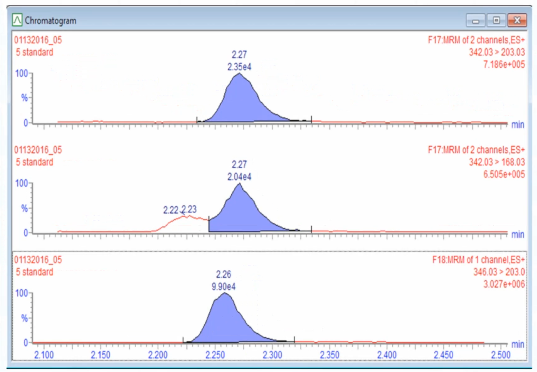 And what you see is that that peak has been there all along. So that was like bad method development on my part that we didn't resolve this peak. But when the plumbing is good, that peak is separated, right. But this is the interesting thing to me like with the UPLC system.
And what you see is that that peak has been there all along. So that was like bad method development on my part that we didn't resolve this peak. But when the plumbing is good, that peak is separated, right. But this is the interesting thing to me like with the UPLC system.
 So this is `the guard column`. And when we took it off, this distance between the end of the ferrule and the end of the tubing just like you were talking in your example, I have to tell you, to me, it looked okay, but it looked a little bit short. And so we redid `the guard column`.
So this is `the guard column`. And when we took it off, this distance between the end of the ferrule and the end of the tubing just like you were talking in your example, I have to tell you, to me, it looked okay, but it looked a little bit short. And so we redid `the guard column`.
 And then the chromatogram, it was like it was a misplaced ferrule and a chromatogram went back to what it was supposed to be. So I think that's one of the lessons I've taken away from working with a UPLC system, it can literally be microliters of difference or millimeters of difference. And you can have bad chromatography. So this is this is the inlet that I was talking about. This is horrible thing where you stick this non wrench there to hold it. And it has a peak ferrule on stainless steel. And so the position of that ferrule makes a big difference. And that one is too short, right, there's a space between the ferrule and the nut. And this one is correct. And that I've seen that much difference really make a smear on the baseline become a peak when you correct it.
And then the chromatogram, it was like it was a misplaced ferrule and a chromatogram went back to what it was supposed to be. So I think that's one of the lessons I've taken away from working with a UPLC system, it can literally be microliters of difference or millimeters of difference. And you can have bad chromatography. So this is this is the inlet that I was talking about. This is horrible thing where you stick this non wrench there to hold it. And it has a peak ferrule on stainless steel. And so the position of that ferrule makes a big difference. And that one is too short, right, there's a space between the ferrule and the nut. And this one is correct. And that I've seen that much difference really make a smear on the baseline become a peak when you correct it.
Misplaced Ferrule #
 So this is my personal favorite photograph. This is the one we talked about. It's from sepscience website. If you Google it, you can find it. So this is what you want to peak connection or in fact a stainless steel connection to look like right the tubing should be butted up right against, they call this the meeting port it's in as far as it can go, it's pretty easy to get it like that it's the first the this hard part is making sure it stays like that while you tighten the fitting down. And here there's a gap, you're never gonna see this gap. But this gap will cause you those kinds of problems that we're seeing, right? That's why it's important to learn how to do fittings correctly.
So this is my personal favorite photograph. This is the one we talked about. It's from sepscience website. If you Google it, you can find it. So this is what you want to peak connection or in fact a stainless steel connection to look like right the tubing should be butted up right against, they call this the meeting port it's in as far as it can go, it's pretty easy to get it like that it's the first the this hard part is making sure it stays like that while you tighten the fitting down. And here there's a gap, you're never gonna see this gap. But this gap will cause you those kinds of problems that we're seeing, right? That's why it's important to learn how to do fittings correctly.
Slide #
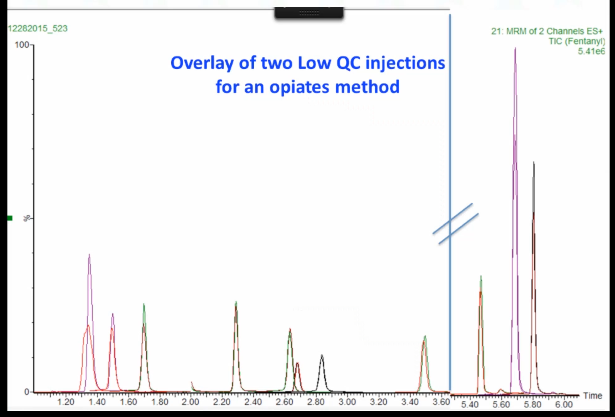 Okay, so the people in our short course saw this one already, so you shouldn't cheat and say. So this is an 11 component opiate method that we had. It is I confessed a dilute and shoot method. And I've truncated there's this big wasted space in here between 3.6 and 5, but I'm showing you all the peaks there in a [composite x I see ??]. You can see something looks a little funny. And this is actually two injections, one from one day and one from the next day that are overlaid on top of each other.
Okay, so the people in our short course saw this one already, so you shouldn't cheat and say. So this is an 11 component opiate method that we had. It is I confessed a dilute and shoot method. And I've truncated there's this big wasted space in here between 3.6 and 5, but I'm showing you all the peaks there in a [composite x I see ??]. You can see something looks a little funny. And this is actually two injections, one from one day and one from the next day that are overlaid on top of each other.
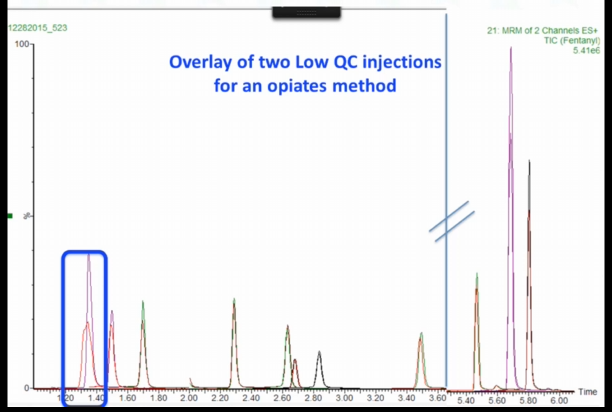
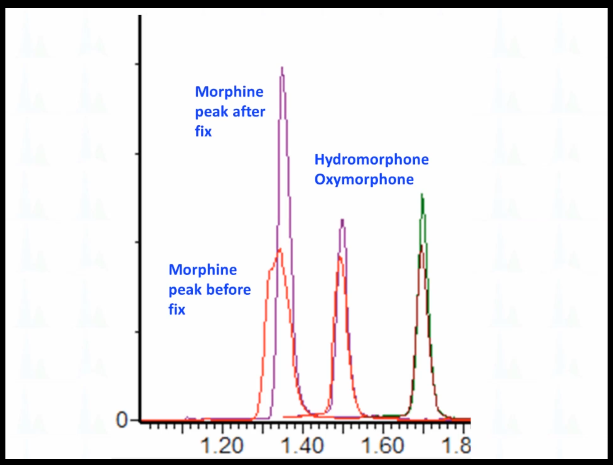 And if we zoom in, this is the earliest saluting peak morphine and this is the problem peak. And that's what it looks Like after we fixed it injecting exactly the same vial. And the next two peaks. This is oxymorphone, and this is hydromorphone. And you can see that although this peak, these are the second injection is a little bit lower, the peak shape is really not affected at all right? It's perfectly overlaid. It's not that wide. So what do you think could be going on here? Why is this? Why does this morphine peak affected strangely and not the rest of them?
So we're kind of in a plumbing theme anyway. What do you think? Do you think this could be a plumbing problem? How many people think this could be a plumbing problem? It's just affecting one peak, the earliest eluting peak. Interesting. Nobody thinks it could be that. The whole batch obviously there's like 30 samples in the batch, all the morphemes looked like this. It actually turned out to be as far as we could tell a worn ferrule.
And if we zoom in, this is the earliest saluting peak morphine and this is the problem peak. And that's what it looks Like after we fixed it injecting exactly the same vial. And the next two peaks. This is oxymorphone, and this is hydromorphone. And you can see that although this peak, these are the second injection is a little bit lower, the peak shape is really not affected at all right? It's perfectly overlaid. It's not that wide. So what do you think could be going on here? Why is this? Why does this morphine peak affected strangely and not the rest of them?
So we're kind of in a plumbing theme anyway. What do you think? Do you think this could be a plumbing problem? How many people think this could be a plumbing problem? It's just affecting one peak, the earliest eluting peak. Interesting. Nobody thinks it could be that. The whole batch obviously there's like 30 samples in the batch, all the morphemes looked like this. It actually turned out to be as far as we could tell a worn ferrule.
 All I can say is that when we looked at it, so this grayness, this is the one we took off, it's just that it's been in touch with the stainless steel, right? It's a little bit my experience the edges of the How would you call that the mouth of the Farrule kind of get thinned out, it looks a little bit more rounded here. So all we did was replaced the peak ferrule, it could have been the position of the ferrule.
All I can say is that when we looked at it, so this grayness, this is the one we took off, it's just that it's been in touch with the stainless steel, right? It's a little bit my experience the edges of the How would you call that the mouth of the Farrule kind of get thinned out, it looks a little bit more rounded here. So all we did was replaced the peak ferrule, it could have been the position of the ferrule.
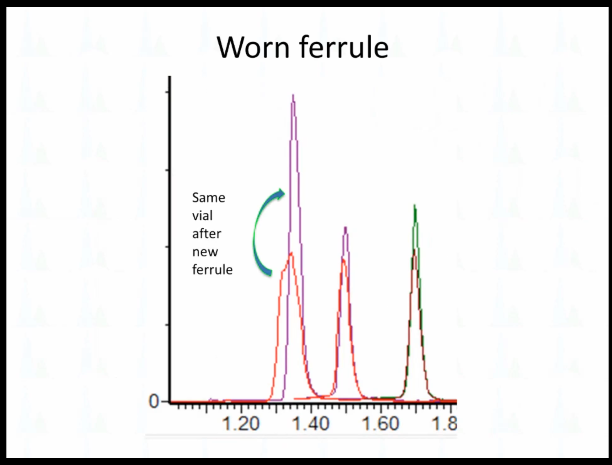 And then it went to that. I personally that's the first time I've ever seen that, that that small of a difference would affect just one peak like that. But that's literally all that happened. We just replaced the ferrule and everything else was good with chromatography. So I think the other lesson that I would take from this is what happened that morning is that somebody had changed `the guard column` here. So we did not change `the guard column`. When we fix it. All we did was change the connection. But I think every time especially with a UPLC system that somebody touches a connection, you have to be suspicious that something might go wrong with it. Has that been your experience? Well, it's that touchy. Yep.
And then it went to that. I personally that's the first time I've ever seen that, that that small of a difference would affect just one peak like that. But that's literally all that happened. We just replaced the ferrule and everything else was good with chromatography. So I think the other lesson that I would take from this is what happened that morning is that somebody had changed `the guard column` here. So we did not change `the guard column`. When we fix it. All we did was change the connection. But I think every time especially with a UPLC system that somebody touches a connection, you have to be suspicious that something might go wrong with it. Has that been your experience? Well, it's that touchy. Yep.
Plumbing #
 Okay, so minimize your extra column volume at the time of the LC install, I would not assume that the vendor is necessarily going to pay that much attention to it. Not everybody cares that much about extra dead volume. So you may have to interact with the service guy to to let him know that's a priority for you. You need to know how to make good connections so that you don't increase the dead volume, you're gonna have to train people, peak fittings don't last forever, they may be pricey, and so it makes you nervous to use it twice and then throw it away, but definitely they age. If you over tighten peak, it's just going to last for a shorter amount of time. If you finger tighten it and then do a quarter turn that should be enough. I would advise you to buy pre-cut stainless steel because it's pretty hard to cut stainless steel on your own and figure out how to get stainless steel ferrule to switch to tubing in the correct length like I was talking about.
Okay, so minimize your extra column volume at the time of the LC install, I would not assume that the vendor is necessarily going to pay that much attention to it. Not everybody cares that much about extra dead volume. So you may have to interact with the service guy to to let him know that's a priority for you. You need to know how to make good connections so that you don't increase the dead volume, you're gonna have to train people, peak fittings don't last forever, they may be pricey, and so it makes you nervous to use it twice and then throw it away, but definitely they age. If you over tighten peak, it's just going to last for a shorter amount of time. If you finger tighten it and then do a quarter turn that should be enough. I would advise you to buy pre-cut stainless steel because it's pretty hard to cut stainless steel on your own and figure out how to get stainless steel ferrule to switch to tubing in the correct length like I was talking about.
Making a good LC connection requires training #
 So for me, what I like to do with the peak fitting is slide it way back on the tubing first. And then when you're putting it into whatever you're connecting to you make sure that the tubing bottoms out because you want the tubing to bottom out and not the fitting. Then you move the fitting forward and you have to hold the tubing in while you're tightening it sometimes that's the hardest part, you need a third hand and then I like to tug on it a little bit to let let me know that it really worked. And remember to replace it. And this is everybody has their own favorite type of tubing cutter. But this is the kind I like how about you? Do you use that kind? The old fashioned ones with like just the actual razor blade in a in an aluminum block? Uh huh. Yeah, but basically, you want to be able to look at the tubing and have a perfectly flat and you don't want an angle you don't want the edges standing up.
So for me, what I like to do with the peak fitting is slide it way back on the tubing first. And then when you're putting it into whatever you're connecting to you make sure that the tubing bottoms out because you want the tubing to bottom out and not the fitting. Then you move the fitting forward and you have to hold the tubing in while you're tightening it sometimes that's the hardest part, you need a third hand and then I like to tug on it a little bit to let let me know that it really worked. And remember to replace it. And this is everybody has their own favorite type of tubing cutter. But this is the kind I like how about you? Do you use that kind? The old fashioned ones with like just the actual razor blade in a in an aluminum block? Uh huh. Yeah, but basically, you want to be able to look at the tubing and have a perfectly flat and you don't want an angle you don't want the edges standing up.
Troubleshooting tools #
 So I'm kind of saying the same thing that well did the maintenance calendar was anything changed how many injections on the column and the guard column. This is the tracking that we talked about. Look at the peak shape of the last run for me if it's slowly degrading, and you can see with our guard column from injection to injection, how it's getting worse, that tells you it's probably a component is aging, if it's sudden that suggests more that it was a bad connection. Also, we unfortunately seem to have a lot of edits made to our methods that aren't supposed to be there so check to see what actually happened if you thought you injected 10 microliters was it really 10 or did somehow was a you know 100 Which actually wouldn't be injected but too large of a volume and of course check this this system suitability test.
So I'm kind of saying the same thing that well did the maintenance calendar was anything changed how many injections on the column and the guard column. This is the tracking that we talked about. Look at the peak shape of the last run for me if it's slowly degrading, and you can see with our guard column from injection to injection, how it's getting worse, that tells you it's probably a component is aging, if it's sudden that suggests more that it was a bad connection. Also, we unfortunately seem to have a lot of edits made to our methods that aren't supposed to be there so check to see what actually happened if you thought you injected 10 microliters was it really 10 or did somehow was a you know 100 Which actually wouldn't be injected but too large of a volume and of course check this this system suitability test.
Column degradation vs bad plumbing? #
 so that's just what I talked about. If you have a lot of injections on the guard comm suspicious that it's degraded if you have zero then probably it's just a connection problem.
so that's just what I talked about. If you have a lot of injections on the guard comm suspicious that it's degraded if you have zero then probably it's just a connection problem.
What is wrong with peaks on the left? #
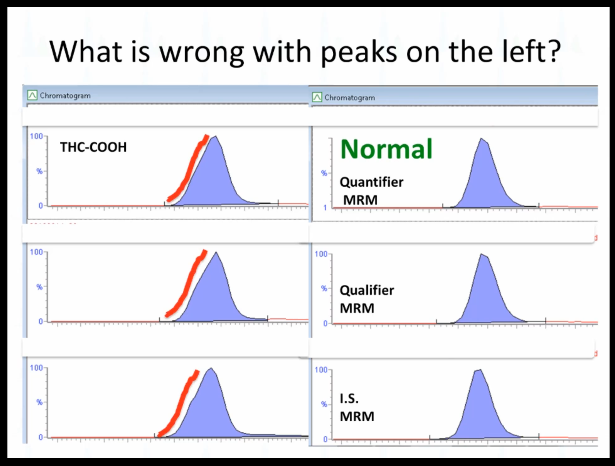 Alright, so we're close to finishing up here. What is What do you think is wrong with these peaks? This is the same same vial essentially sorry, it's not true. This is the same standard injected here versus here. What don't you like about this side? What is wrong with these peaks fronting is everybody know what fronting means? That means this here, it's not symmetrical. Okay, it's kind of subtle, if you had all of them look like that, you might just find out that you, that's how you design the asset, but it really should look more symmetrical.
Alright, so we're close to finishing up here. What is What do you think is wrong with these peaks? This is the same same vial essentially sorry, it's not true. This is the same standard injected here versus here. What don't you like about this side? What is wrong with these peaks fronting is everybody know what fronting means? That means this here, it's not symmetrical. Okay, it's kind of subtle, if you had all of them look like that, you might just find out that you, that's how you design the asset, but it really should look more symmetrical.
Wrong injection matrix (columm overload) #
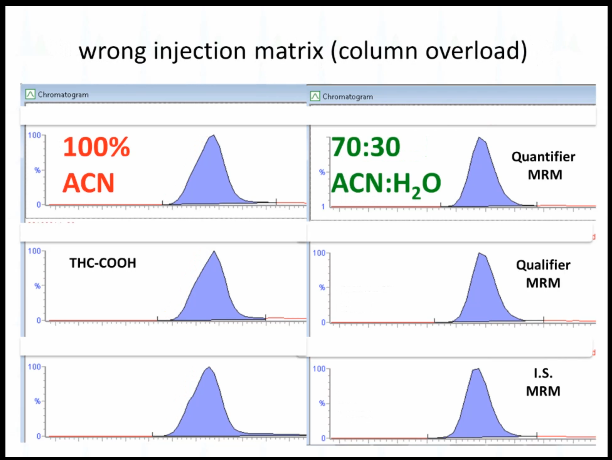 And so it was the wrong injection matrix. This is a solid phase extraction for marijuana metabolite. it's supposed to be reconstituted with 70:30 acetonitrile:Water. Instead, it was reconstituted with 100% of acetonitrile. Okay, so in 100%, acetonitrile, essentially, the THC kind of likes the mobile phase more, that's how I think of it. And so it doesn't adhere in a tight band to the head of the column. So we had to re extract the whole batch, and then it was okay.
And so it was the wrong injection matrix. This is a solid phase extraction for marijuana metabolite. it's supposed to be reconstituted with 70:30 acetonitrile:Water. Instead, it was reconstituted with 100% of acetonitrile. Okay, so in 100%, acetonitrile, essentially, the THC kind of likes the mobile phase more, that's how I think of it. And so it doesn't adhere in a tight band to the head of the column. So we had to re extract the whole batch, and then it was okay.
Wrong injection matrix #
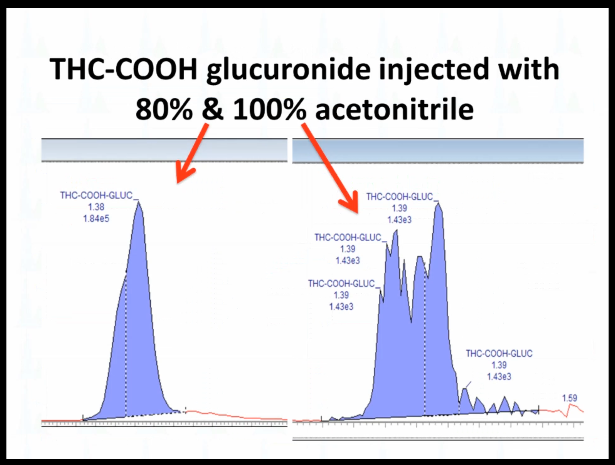 If you're looking at something that's more nonpolar, like THC-COOH glucuronide, we're looking at the free acid metabolite there. It can get this kind of a fronting or even this, this is how bad it looks, you don't even get a peak, you just get a blob, when the percentage of acetonitrile is too high.
If you're looking at something that's more nonpolar, like THC-COOH glucuronide, we're looking at the free acid metabolite there. It can get this kind of a fronting or even this, this is how bad it looks, you don't even get a peak, you just get a blob, when the percentage of acetonitrile is too high.
What is column overload? #
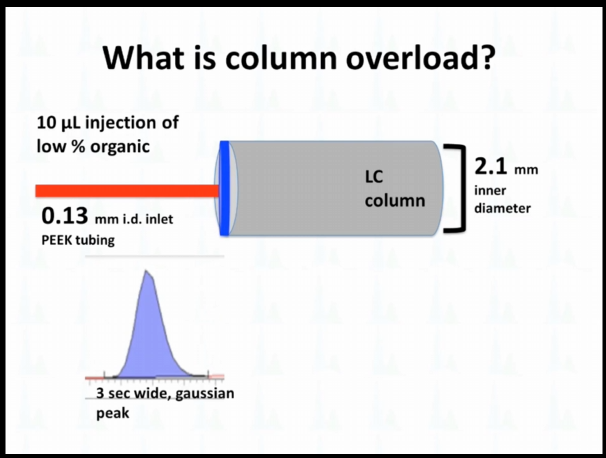
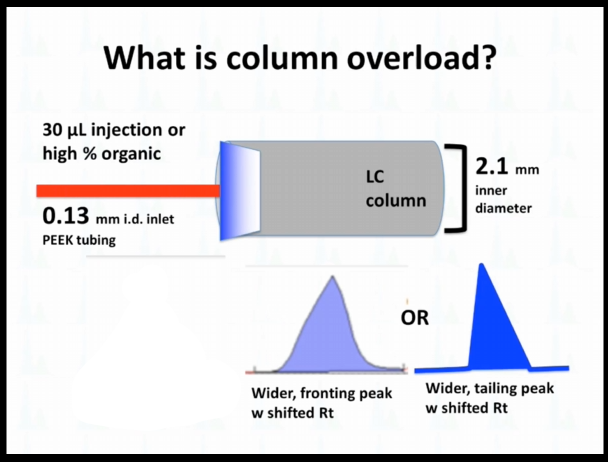 So sometimes I've talked to people at column overload, and I just wanted to they're not so sure what I'm talking about. I just wanted to illustrate that a little bit. So if we do it, right, if we inject the right volume at the right, organic, we get this tight band, and then it forms a nice narrow Gaussian peak. If we're going to inject too much volume, or if it's too high of a percent organic, again, we're talking reverse phase is our default, we're going to see something like this fronting peaks, or wider tailing peak that sometimes has happened.
So sometimes I've talked to people at column overload, and I just wanted to they're not so sure what I'm talking about. I just wanted to illustrate that a little bit. So if we do it, right, if we inject the right volume at the right, organic, we get this tight band, and then it forms a nice narrow Gaussian peak. If we're going to inject too much volume, or if it's too high of a percent organic, again, we're talking reverse phase is our default, we're going to see something like this fronting peaks, or wider tailing peak that sometimes has happened.
Bad Design of LC+Sample Prep #
 And to me, this is really just bad design of your LLC and sample prep. So I think the I'm just going to step through these quickly, I'll post these on the on the website as well. But my experience is that with a gradient analysis, you can't just say, this is going to be too much organic, you really have to take into account your starting conditions on the gradient, your stationary phase, the inner diameter of the column, how much volume you're injecting, and even your analyte.
And to me, this is really just bad design of your LLC and sample prep. So I think the I'm just going to step through these quickly, I'll post these on the on the website as well. But my experience is that with a gradient analysis, you can't just say, this is going to be too much organic, you really have to take into account your starting conditions on the gradient, your stationary phase, the inner diameter of the column, how much volume you're injecting, and even your analyte.
Sample Prep faux pas #
 Because I've been able to inject like 70% organic or 75%, organic, onto a short column with a 2.1 millimeter internal diameter with a starting at like 30% B and I get a very nice tight peak because I really nonpolar, analyte, right, it really likes the stationary phase a lot better than it likes the mobile phase. And so you can get away with that. So I've often said people say, Oh, no, you could never do that. I personally feel that with gradient analysis, you should just try it, you just have to establish it empirically is that your experience? that you can't predict it. And the issue mean, of course, that you want more organic, because you want to get off, and you want to get your compound back off of the glass, or you have better solubility and more organic, but if the organic is too high, then you're going to get bad looking peaks.
Because I've been able to inject like 70% organic or 75%, organic, onto a short column with a 2.1 millimeter internal diameter with a starting at like 30% B and I get a very nice tight peak because I really nonpolar, analyte, right, it really likes the stationary phase a lot better than it likes the mobile phase. And so you can get away with that. So I've often said people say, Oh, no, you could never do that. I personally feel that with gradient analysis, you should just try it, you just have to establish it empirically is that your experience? that you can't predict it. And the issue mean, of course, that you want more organic, because you want to get off, and you want to get your compound back off of the glass, or you have better solubility and more organic, but if the organic is too high, then you're going to get bad looking peaks.
Mobile phase #
 So there are some rules for this, your goal is focusing on the analyte in a narrow band, if you have isocratic versus a gradient, that you have to take that into account.
So there are some rules for this, your goal is focusing on the analyte in a narrow band, if you have isocratic versus a gradient, that you have to take that into account.
Injection matrix “rules” #
 So for isocratic, Russ Grant rule is an injection matrix of organic solvent that's less than or equal to 90% of the mobile phase organic percent. So if you're running at 40%, acetonitrile, then you want your injection matrix to be no more than what is that 36%. And as I said, for gradient, I encourage you strongly not to say Oh, I couldn't possibly I'm starting at 30% V, I couldn't possibly inject anything more than 30% V, I think you absolutely can do that. And potentially give yourself a lot more flexibility.
So for isocratic, Russ Grant rule is an injection matrix of organic solvent that's less than or equal to 90% of the mobile phase organic percent. So if you're running at 40%, acetonitrile, then you want your injection matrix to be no more than what is that 36%. And as I said, for gradient, I encourage you strongly not to say Oh, I couldn't possibly I'm starting at 30% V, I couldn't possibly inject anything more than 30% V, I think you absolutely can do that. And potentially give yourself a lot more flexibility.
LC and Sample Prep Related Peak Shape problems #
 So what we talked about was column guard comm or online filter aging, and a filter can give you that same type of problems, not just the guard, or that you changed it and made a bad connection that you had new tubing or fitting installed post column. And that can also cause a problem wrong injection solvent or the wrong mobile phase, needle phase or column.
So what we talked about was column guard comm or online filter aging, and a filter can give you that same type of problems, not just the guard, or that you changed it and made a bad connection that you had new tubing or fitting installed post column. And that can also cause a problem wrong injection solvent or the wrong mobile phase, needle phase or column.
So that’s about it. That’s a Picasso in case you didn’t recognize it and thanks for hanging in there into your lunch hour or whatever. Thanks very much.
Reference: