Author: Judy Stone, PhD, MT(ASCP) // Date: AUG.1.2015 // Source: Clinical Laboratory News
Summary:
What are problems?
Most laboratories encounter much more LC failures than MS/MS problems, but they make more effort to recover MS/MS sensitivity.
it is the unexpected instrument failure that compromises productivity more than the regularly scheduled preventative maintenance. Therefore, successful troubleshooting skills are vital to diagnose instrument decline early (proactive maintenance) and intervene before there is a batch failure.
List of problems
- signal-to-noise
- retention time shift
- missing peak
- MS/MS sensitivity
- peak abundance
- resolution
- leak
- over pressure
What are good practices of maintenance?
a) Have spare, clean, MS/MS interface parts ready to install
b) Track column and lot changes for as many chemicals, reagents, and solvents as possible.
c) avoid using plastic containers and parafilm
d) ?? practices of adding and discarding residual mobile phase
What is troubleshooting strategy? or What is proactive maintenance about?
- Setup and support proactive maintenance infrastructure
a) injection of neat standards, double blanks, method blanks daily.
b) post-column syringe infusions for the sensitivity to the MS/MS.
c) have a reference point to compare with. The reference could be: baseline, retention time, EIC (extracted ion chromatogram), peak height
d) Good records (Shiny app)
e)
f) - Divide and conquer
a) ‘false alarms’ from analyst failure.
b) ‘Sample preparation’, ‘LC’, ‘MS/MS’
1 mg/dL = 1000 μg/100mL = 10 μg/mL = 1000 x 1000 x 10 pg/mL
Troubleshooting liquid chromatography-tandem mass spectrometry (LC-MS/MS) systems can be intimidating because the technique is complex, instrument operation and sample processing are still quite manual, and the majority of assays are laboratory-developed tests (LDTs). Vendor service and support has improved, but it is unrealistic to expect that a vendor can have the same degree of understanding for an LDT as for a Food and Drug Administration (FDA)–approved test.
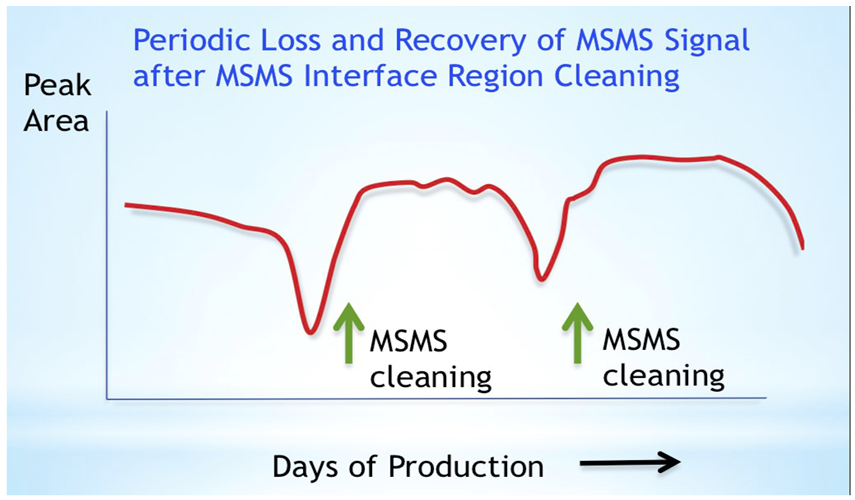
Another challenge is the natural history of LC columns and MS/MS instruments. Chromatography and MS/MS response degrade incrementally with every injection, as residual matrix from extracted sample deposits on the LC column and the MS/MS interface region. Labs replace the LC column when it becomes unusable, and clean the interface region when MS/MS sensitivity falls too low. This pattern recurs regularly; the only variable is the length of time that the MS/MS can operate before cleaning is needed, called the maintenance-free interval. Although most laboratories find LC failures much more often than MS/MS problems, it is often faster to diagnose the root cause and return the LC to normal operations than it is to recover MS/MS sensitivity (Figure 1 1).
The key to minimizing instrument unavailability and producing reliable patient results is implementing sample preparation and maintenance protocols that deliver predictable maintenance-free intervals. Although needing to clean the MS/MS less often does mean that more samples can be tested per instrument, it is the unexpected instrument failure that compromises productivity more than the regularly scheduled preventative maintenance. Therefore, successful troubleshooting skills are vital to diagnose instrument decline early and intervene before there is a batch failure. Of course, troubleshooting will be of little value unless the laboratory knows its instrument well (Table 1 2).
I recommend the following steps to enhance your troubleshooting abilities, outlined below:
- implement a robust production infrastructure,
- build troubleshooting skills, and
- divide and conquer problems to reduce complexity.
1. Set Up and Support a Robust Production Infrastructure
1) Create detailed maintenance charts — with practical language — and insist on complete documentation, including data-derived action limits and corrective actions. Post images to make compliance easier (Table 2 3).
2) Incorporate a System Suitability Test (SST) in daily maintenance. An SST is the injection of neat standards to check LC and MS/MS reagents, parameters, and status (Table 3 4). Think of the SST like a patient’s vital signs—pulse, blood pressure, temperature—for evaluating the health of your LC-MS/MS.
3) Derive realistic practices for adding and discarding residual mobile phase, for changing mobile phase and autosampler wash containers and filters, and for cleaning and storing the containers. Avoiding contamination when performing trace MS/MS analysis at pg/mL concentrations requires a different mindset compared to practices appropriate for the mg/dL concentrations tested on automated, routine chemistry analyzers.
4) Have spare, clean, MS/MS interface parts ready to install. When the MS/MS interface needs cleaning, this decreases from hours to minutes the length of time the instrument is vented to atmospheric pressure, and as a result, less time elapses until the MS/MS is pumped down again to operating vacuum pressure (Figure 2 5).
5) Track column and lot changes for as many chemicals, reagents, and solvents as possible, and avoid using plastic containers and parafilm.
2. Strap on Your Troubleshooting Tool Belt
1) If signal to noise is low, increased noise as well as decreased signal may be a contributor. Compare the baseline to your archived image: elevated baselines suggest contamination of mobile phases, mobile phase containers, or reagents.
2) If peaks are missing or retention time (Rt) shifts, compare pressure traces to archived images — a rapid way to detect LC leaks, overpressure, and pump problems.
3) For almost any problem, review the SST results. The SST can distinguish between an instrument problem versus a sample preparation failure because the sample preparation phase is bypassed by using neat standards.
4) Compare composite extracted ion chromatogram (XIC) overlays from questionable injections to your archived images. Changes in the pattern of relative peak abundance, Rt, or resolution may be immediately apparent with XIC overlays when review of individual peaks is unrevealing.
5) When you question your MS/MS response, compare peak heights from post-column syringe infusions to your historical values to isolate a loss of sensitivity to the MS/MS rather than LC. However, infusion abundance sometimes appears acceptable even when the MS/MS needs service, so always evaluate infusions in the context of the SST.
6) Peruse the maintenance chart and accept that every human intervention (including your own) is suspect when there is a subsequent change in instrument performance. As long as good records are kept, you can appear to be a genius with the quick fix of an operator mistake or non-compliance with the standard operating procedure.
7) Ask the best vendor service representative to teach you his or her protocol for detecting leaks and finding the source of overpressure.
3. Divide and Conquer
1) First rule out false alarms from common mistakes. For example, an error yields bad data, such as injecting the wrong vial ([Table 4 6).
Any maintenance, component, or reagent replacement is susceptible to human error. Don’t hesitate to check, reconnect, repeat, and replace even though it may feel insulting or redundant.
2) Distinguish between sample preparation, LC, and MS/MS problems using the SST and MS/MS infusion.
i) Sample Prep:
a) a normal SST indicates a sample preparation problem, but casual inspection of the SST can lead to a false conclusion that the instrument is working correctly.
b) Evaluate SST results for trends over time as well as for out-of-range values.
c) Verify that the vial or plate cap was pierced by the autosampler needle and confirm that the liquid contents appear as expected.
d) Re-inject a stable extracted sample from a previous batch to confirm acceptable instrument performance.
e) Also, check for lot changes.
f) Finally, a step-by-step review with the analyst who performed the sample preparation may be the only remaining option before repeating the extractions (See Table 5 for a list of common mistakes categorized by type of sample preparation).
ii) MS/MS:
If infusion of standards or calibration solutions indicates the MS/MS signal is decreased, first rule out interventions that do not require venting. For example, confirm that the MS/MS detector voltage is appropriate, mass resolution and calibration are correct, and no ion source maintenance (e.g. new capillary electrode) is needed. Consulting the pattern of MS/MS maintenance-free intervals for the instrument may be helpful (SST records), although one bad sample or a change in the assays performed on the instrument can cause a dramatic loss of response. Most clinical laboratories consult vendor technical support and request onsite service at this stage, although competent end users can change interface components followed by mass resolution and calibration.
iii) LC:
LC-related problems are the most common reason for SST and batch failures. Review of the SST report, XIC overlays, and pressure traces are the best tools to detect and solve an LC problem. If you suspect a leak, look at and touch every tubing connection from the pump to the MS/MS source while the instrument is pumping, and look for buffer deposits below a connection or discoloration of metal fittings (green) as indication of a slow leak. Leaks often occur following over pressure.
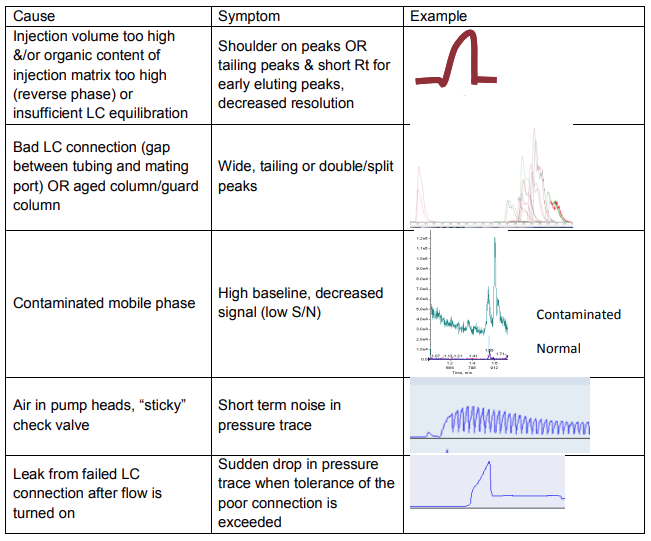
3) Selected distortions in peak shape and pressure traces are diagnostic for specific injection matrix, LC plumbing, LC pump, and column problems (Table 6 7).
Even if you can’t solve the problem, taking the first steps and sharing your troubleshooting data with the vendor service representative can get the instrument back in to service more quickly. Discussions with and learning from the service representative also boosts your troubleshooting skills, although nothing is more rewarding than finding the root cause of an obscure LC problem on your own. Grab your wrenches and troubleshoot!
Table 5 Some common sample preparation problems by extraction type
Type of Sample Preparation |
Common Problems and Outcomes |
|---|---|
Solid Phase Extraction (SPE) |
Elute analytes to waste instead of autosampler vials or collection plate – no or low peak areas |
Solid Phase Extraction (SPE) |
Silica based SPE stationary phase dries out during extraction (selected cartridges/wells) – low, inconsistent peak areas. Polymer based SPE media is not sensitive to drying out. |
Liquid-Liquid Extraction |
Contamination of organic layer with the interface between aqueous/organic layers during transfer – interfering peaks, ion suppression, low peak areas, LC over pressure from injected particulate |
Liquid-Liquid Extraction |
Emulsion – no or low peak areas, extraction may need to be repeated if unable to break emulsion |
Protein precipitation with filtration plates or phospholipid removal plates |
Clogging of selected wells, no filtration – no peaks, no liquid in well of collection plate |
Manual protein precipitation |
Insufficient precipitation, delayed precipitation in autosampler vial, insufficient mixing, insufficient centrifugation - LC overpressure from injected particulate |
Protein precipitation (serum, plasma), Simple dilution (urine, CSF) |
Insufficient matrix removal – variable ion suppression, shortened column lifetimes, shortened MS/MS maintenance-free intervals |
Reference: AACC: Troubleshooting Liquid Chromatography-Tandem Mass Spectrometry in the Clinical Laboratory
-
Figure 1
Extracted biological fluid samples contain varying degrees of residual matrix which deposits on the front end components of the MS/MS (entrance plate and interface region) when samples are injected on the LC-MS/MS system. Eventually the interface region becomes contaminated and MS/MS performance is compromised—for example sensitivity (peak area) is decreased. The MS/MS is vented to atmospheric pressure and cooled. Interface components are removed and cleaned or replaced. The MS/MS is pumped down to operating vacuum pressures (e.g. 1 e-5 torr) and returned to use after checking/updating mass resolution and calibration. Typically the instrument is out of service for 18-24 hrs, most of the time spent on the pump down phase.
 ↩︎
↩︎ -
Table 1 Know your instrument better than you know your co-workers
1. Learn the LC flow path.
2. Practice cutting the PEEK tubing that connects LC components and making good connections with PEEK and stainless steel tubing/fittings. Be able to recognize a good versus a bad tubing cut and connection.
3. Know how to find the instrument method parameters stored with every acquired LC-MS/MS data file and archive an example when each assay is validated.
4. Every autosampler model is somewhat different. Learn how your injector introduces samples and is washed to prevent carryover. Know the sample loop size.
5. Archive screen shots of normal LC pressure traces.
6. Archive screen shots of normal baseline for each method.
7. Create and archive composite extracted ion chromatogram (XIC) overlays with a fixed Y scale for all MRMs in a low calibrator with good chromatography. This documents normal retention times (Rt), peak shapes, resolution and relative heights.
8. As a baseline for future reference record the MS/MS response on more than one occasion from post-column infusion of pure standards in methanol. Write a protocol including the instrument parameters used for infusion.
9. Archive photographs of the MS/MS ion source and the cone, curtain plate or skimmer (differs by vendor - the first heated MS/MS surface encountered by volatilized samples) when clean. Some discoloration of this metal surface is expected, but deposition of excess salts and carbonized matrix may indicate errors in maintenance, divert valve editing, reagent or sample preparation. ↩︎ -
Table 2 Recommendations for items to include in an LC-MS/MS maintenance calendar
Customize as needed based on vendor guidance, number of samples tested, extent of sample clean-up (minimal or extensive matrix removal during extraction)
Daily System Checks & Instrument Preparation
1. Sufficient mobile phases & autosampler wash
2. Empty liquid waste
3. Prime mobile phases & auto-sampler wash
4. Record MS/MS vacuum pressures, verify in range
5. Sufficient MS/MS gas (N2/Ar) inlet pressure
6. Record # of injections on columns & guard columns, change as indicated
7. Run SST & record results, compare to action limits
8. Record LC pressure during SST, compare to action limits
9. Check LC for leaks
Periodic Checks (daily, weekly, monthly, as needed)
1. Roughing pump oil level, ballast pump
2. Change filters in mobile phase, autosampler wash solutions
3. PC maintenance (reboot, disk cleanup & defragmentation, OS updates). Backup/delete data files
4. Clean cones (curtain plate, skimmer, skimmer plate)
5. Check MS/MS resolution, calibration & response with infusion of a standard, compare to action limits
6. Perform mass resolution & mass calibration following service or with frequency per vendor recommendations
Best Practices
1. Change mobile phase bottles frequently, avoid adding mobile phase to existing bottle when possible, follow guidelines to miminize mobile phase and container contamination.
2. After the run, wash columns and leave LC system in 100% organic solvent (e.g. acetonitrile) with no acid, no buffer.
3. Record all parts, tubing, fitting column, filter replacements
4. Autosampler, pump, flow switching valve preventative maintenance as needed with high volume testing (needle, needle seal, rotor seal, plunger seal, check valves)
5. Track solvent, chemical lot#s ↩︎ -
Table 3 System Suitability Test Outline
Protocol
1. 3-5 injections of pure standard(s) in injection matrix (and internal standards if desired)
2. Record mean and %CV of Rt, peak area (peak height, peak width)
3. Compare means & %CVs to action limits
Recommendations
1. Prepare standard solutions in injection matrix in bulk, aliquot to autosampler vials and store ready for use at -200C (or best temperature for stability).
2. Graph peak area most sensitive to MS/MS status and evaluate for trends.
Options
1. Verify acceptable separation of peaks most sensitive to column degradation (if applicable).
2. Document (screen shot, print) acceptable peak shape for analyte(s) most sensitive to column degradation.
3. Verify baseline signal (counts per sec or cps) is below action limit ↩︎ -
Figure 2
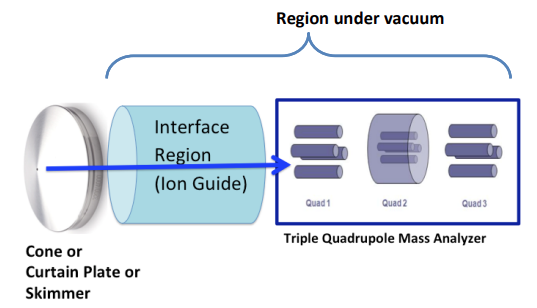
The MS/MS “interface” is defined generically as the hardware components under vacuum between the ion source and the first mass analyzer quadrupole. These components differ in design from one vendor to another (IonDrive™, iFunnel , Stepwave Ion Guide, Active Ion Management [AIM™]) but have the common purpose of increasing ion transfer efficiency and reducing transfer of neutral species and gases from the ion source to the mass analyzer region. These surfaces receive the maximum exposure to residual matrix from extracted biological fluids and environmental contaminants, and therefore are susceptible to contamination and compromised function. The blue arrow denotes the direction of the ion path.
 ↩︎
↩︎ -
Table 4 Common “false alarm” mistakes – operator failure instead of instrument failure
1. Injecting the wrong vial (mistaken position in autosampler tray) for SST – no or wrong peaks
2. Injecting the wrong volume (typically 0 volume) for SST – no peaks or low peak areas
3. Insufficient liquid in vials for SST – high %CV with latter injections having low peak area, no peak
4. Insufficient equilibration of LC column before running SST – no peaks and/or shifted Rt, bad peak shapes, increased peak widths, poor resolution - particularly for early eluting analytes
5. Mobile phase runs out during SST – no peaks or shifted Rt, low LC pressure
6. Autosampler wash runs out during SST – low peak areas, carryover
7. Ion source not reconnected or valve not directed to MS/MS after cleaning cones (curtain plate, skimmer, skimmer plate) – no peaks, no baseline (except electronic noise)
8. Wrong acquisition method used for SST – no or wrong peaks
9. Wrong data analysis method used for SST – no peaks, selected peaks are missing
10. Data files stored in wrong project for SST – no peaks, no baseline ↩︎ -
Table 6 Examples of LC problems diagnosed from peak shape and pressure traces
 ↩︎
↩︎