Tips: Frequently, resolving the problem requires a combination of individual experimental solutions and systematic and logical investigation.
Part 1 highlights rational method development and validation and includes troubleshooting tips for sound sample preparation, for analyte stability in biological matrices, and for chromatographic conditions and injection solvents.
Part 2 further discusses development strategies for LC/MS/MS analysis of biopharmaceutical products including proteins and peptides. It will present troubleshooting tips for carryover, recovery and matrix effects, and selectivity; and for automation where possible to improve the overall reliability and efficiency of LC/MS/MS.
Part 1 #
Sample Preparation #
(1) The first step in sample clean-up is to remove as many of the unwanted compounds (including macromolecules and other compounds such as proteins, endogenous and exogenous compounds, and coadministered pharmaceuticals) as possible without significant loss of the analytes of interest. Solid-phase extraction (SPE), liquid–liquid (LL) extraction, and protein precipitation (PP) are often the techniques of choice. Unwanted compounds can be present still in higher concentrations than the analytes of interest after the first clean-up. (2) A second stage of clean-up, typically involving LC separation further separates analytes of interest from the unwanted compounds. (3) MS/MS offers a third stage of separation through selection of appropriate precursor and product ion pairs so that unwanted compounds are not registered (unseen) by the detector.
However, those unwanted and MS/MS unseen compounds present significant challenges for LC/MS/MS practitioners. In the LC/MS interface, unwanted compounds compete with analytes for ionization and can cause inconsistent matrix effects that are detrimental to quantitative LC/MS/MS. If not separated from the analytes, some conjugated metabolites break down in the interface so that analyte concentration is overestimated. LC/MS/MS practitioners must use caution in their analyses and envision that behind every analyte peak, “unseen” MS peaks from contaminants may cause or contribute to potential problems in the assessment.
Analyte stability #
Certain drug product analytes are subject to degradation. Endogenous enzymes found within biological matrices can accelerate analyte degradation. Information about degraded analytes should be obtained during method development. A general troubleshooting approach to address that problem would be stabilizing analytes by choosing an appropriate anticoagulant, pH, or enzyme inhibitor. If analytes cannot be stabilized, then in situ derivatization of the analytes to more stable forms in biological matrices should be attempted.
Chromatographic Conditions #
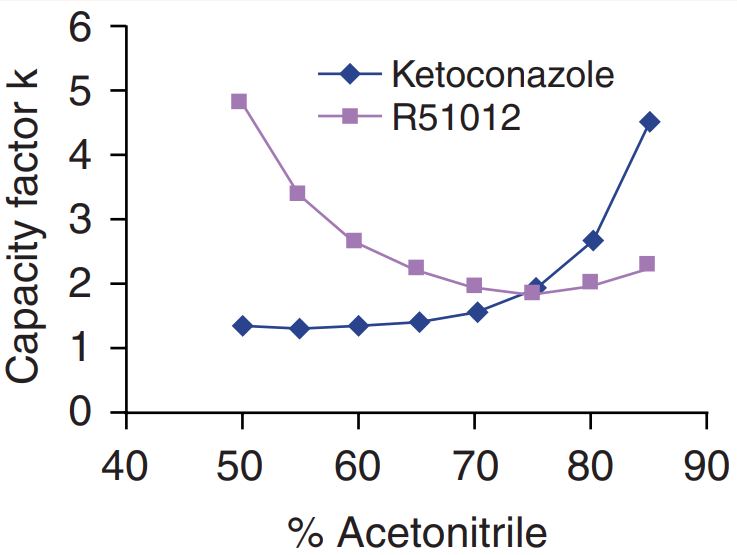
On-column retention. Reversed-phase columns do not always behave in a “reversed-phase” way. The characteristics of the analyte, the column, and the mobile phase can interact with unexpected results. For example, Figure 1 shows that when organic content of the mobile phase composition was increased (a desirable characteristic for improving sensitivity), the capacity factor (k') decreased initially but increased after the deflection point. That system demonstrated an initial reversed-phase mode with primary retention because of hydrophobic interaction between the analytes and the alkyl chains. It reverted to normal-phase where the primary retention was because of hydrophilic interaction between the analyte and the residual silanol groups. That kind of dual retention behavior was observed for various analytes on different brands of reversed-phase columns. Even the extensively end-capped reversed-phase column had at least 30% nonendcapped residual silanol groups. Analyte on-column retention, therefore, depends on the characteristics and interactions of the analyte, the column, and the mobile phase.
Polar ionic retention. For analysis of polar ionic compounds, reversed-phase LC/MS/MS can be problematic. Anion formation occurs when the pH is higher than the pKa in acidic compounds, and cation formation occurs when the pH is lower than the pKa of basic compounds. Column retention decreases matrix effects. Ionization of polar analytes decreases columnretention, and promotes matrix effects that can impact analysis bioanalysis. To retain polar ionic compounds, highly aqueous mobile phases or ion-pair chromatography is required. However, neither of those methods is conducive to the spray conditions required to achieve good LC/MS/MS sensitivity. Good spray conditions are necessary for strong, stable analyte signals.
A number of drugs have basic functional groups, and MS in the positive ion mode often detects such components as protonated ions. Protonation is the most important means of ionization in positive-ion electrospray mass spectrometry. Acidic mobile phases often are used to ensure that analytes are in their protonated forms. However, the charged analytes will have poorer retention on reversed-phase columns. That is undesirable because detrimental matrix effects can obscure the analysis.
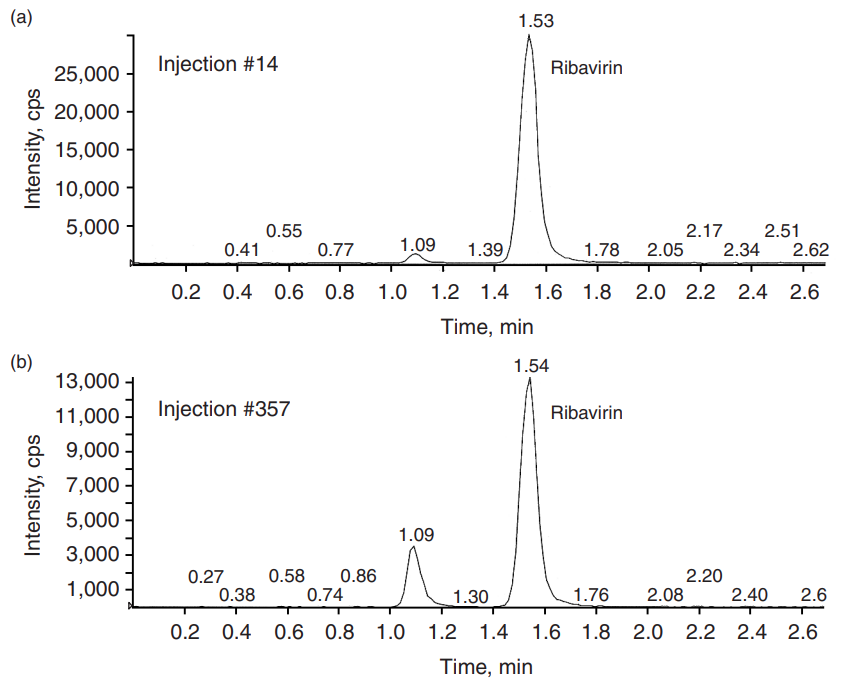
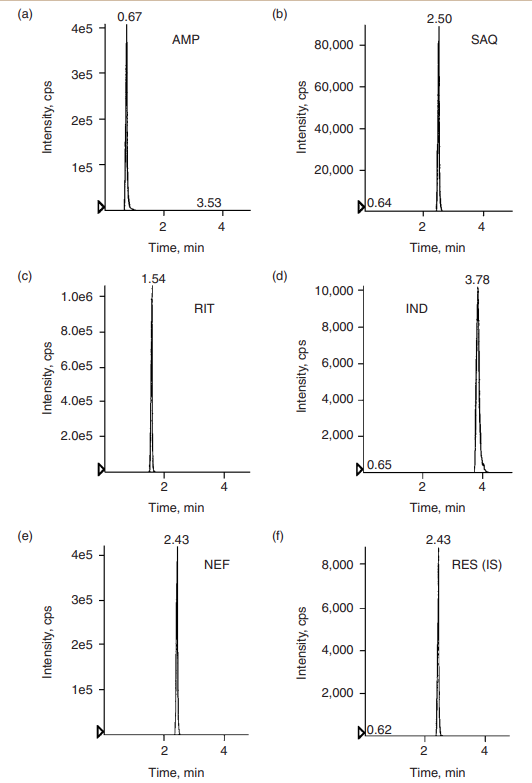
Silica columns demonstrate excellent stability. Typically, one column can be used for at least 500 injections of extracted samples. Figure 2 shows column stability of the LC/MS/MS analysis of ribavirin in human plasma after 350 injections. Even with the protein precipitation extraction method, no retention-time changes were observed. The combination of an aqueousorganic mobile phase and a silica column also demonstrated an excellent, reproducible gradient elution. Reequilibration time was comparable with that of the reversed-phase column. Table 2 lists retention times for four consecutive injections of five different protease inhibitors. The resulting chromatograms demonstrate excellent peak shape and resolution, shown in Figure 3.
Injection Solvents #
A trend toward faster, more efficient LC/MS/MS has prompted the use of columns with smaller dimensions (50 μm x 2 μm). In contrast to conventional columns (250 μm x 4.6 μm), one column offers advantages that include faster analysis time, better concentration sensitivity, and lower solvent consumption. However, when smaller quantities of packing material are used, the chromatography can be more easily disturbed. Mismatches between the injection solution and the mobile phase are a common problem in compounds that demonstrate early elution peaks (21).
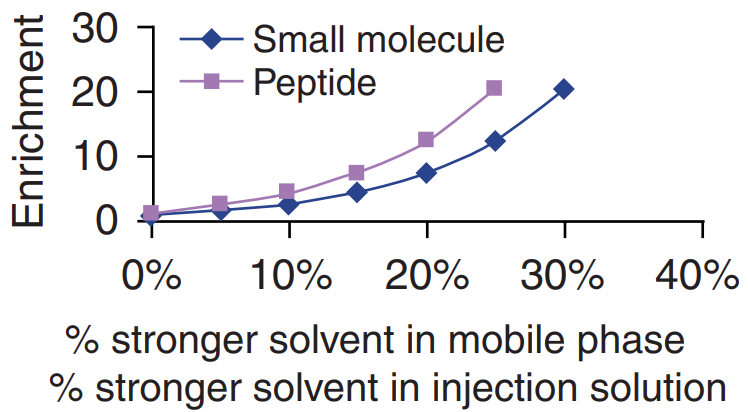
Choosing a reconstitution solvent compatible with the mobile phase can increase sensitivity. When the injection solution has a weaker eluting strength than the mobile phase, the effects of the injection solution on peak shape and chromatography efficiency can be enriched. Figure 4 shows a typical enrichment profile. Maximum chromatography efficiency can be achieved using injection solution at the weakest elution strength. From a practical point of view, better chromatography efficiency can be obtained by diluting the sample with the weaker component in the mobile phase and increasing the injection volume.
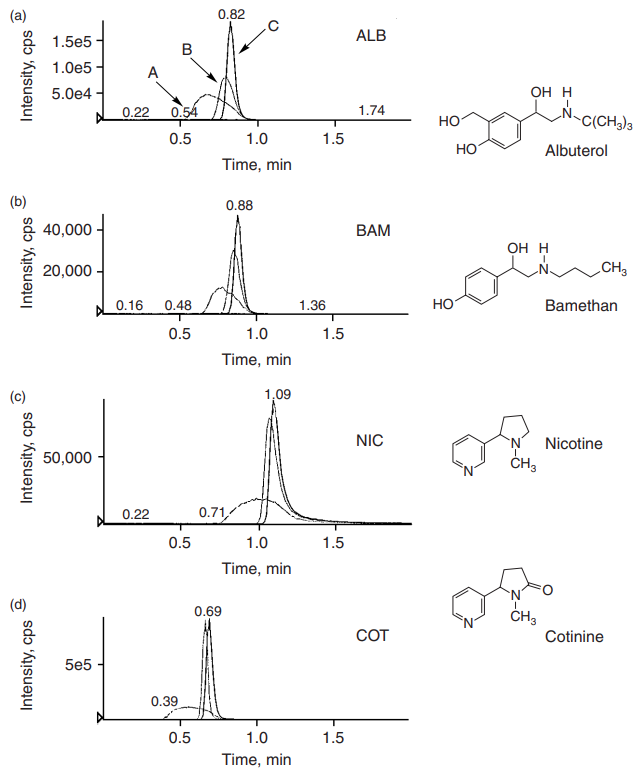
C18 columns. Before choosing a diluting solvent, you should determine the retention mechanism. Retention mechanisms dictate whether water or organic solvent will be stronger for elution. For reversed-phase LC, water is weaker than organic solvents, such as acetonitrile, are stronger. For normal phase LC, the solvent strengths are reversed: Water is the stronger elution solvent, and organics the weaker. Even the name “reversed-phase C18 column” can be misleading. The retention mechanism on a C18 column can be complicated by the dual retention mechanism, as shown in Figure 1. The actual retention mechanism depends on the analyte itself, the mobile phase composition, and the stationary phase. If retention on the C18 column were blindly assumed to be reversed-phase, water would be used as the weaker elution solvent and, therefore, the injection solution. Very poor peak shapes would result. The influence of injection solvents on chromatography is demonstrated in Figure 5.
If an analyte is unstable in the mobile phase but stable in a weaker component of it, the mobile phase can still be used, as long as the weaker solvent is used for reconstitution. For example, omeprazole is unstable in acidic solution. To obtain good peak shape and sensitivity, an acidic mobile phase can be used. After extraction, omeprazole can be reconstituted in acetonitrile and injected onto a silica column with an acidic aqueous–organic mobile phase (13). Because of the short run time (three minutes), on-column degradation of omeprazole is not observed. In that situation, both analyte stability and on-column stacking are achieved because acetonitrile is a weaker solvent for the silica column (see the “Injection Solution Selection Criteria” box).
Reference: Systematic Troubleshooting for LC/MS/MS - Part 1: Sample Preparation and Chromatography
Part 2 #
Carry-over #
Carry-over is probably one of the most commonly encountered problems of LC/MS/MS method development. Sources of problems range from instrument hardware and the selection of appropriate rinse solvents to challenges with chromatography. Frequently, resolving the problem requires a combination of individual experimental solutions and systematic and logical investigation.
The gradient elution was actually determined to be the cause of the carry-over effect. To address the issue, (1) an isocratic elution with a mobile phase that contained 45% acetonitrile was used to eliminate the carryover. A new problem surfaced when the original internal standard was not retained on the column in the mobile phase, so a new one had to be used. The new standard had retention properties similar to those of the analyte. Figure 2 depicts the resolution of the carry-over problem. The method was validated and successfully used for sample analysis. chromatographic condition and internal standard were changed as a result of the problem and its solution. Occasionally, carry-over problems have simpler solutions. In another example, simply raising the needle height solved the carry-over effect. (2) Carry-over is often avoided by merely minimizing the contact surface between analyte and needle.
Occasionally, carry-over problems have simpler solutions. In another example, simply raising the needle height solved the carry-over effect. (3) Carry-over is often avoided by merely minimizing the contact surface between analyte and needle.
Recovery and matrix effects #
For an LC/MS/MS method, it is important to identify whether the loss of sensitivity is due to poor recovery or to matrix suppression, because both causes give the same result. Carefully designed experiments will establish the source of the problem.
Recovery is determined by comparing the peak areas of extracted samples with those of neat solutions spiked (postextraction) into a blank matrix. Because both samples have the matrix ingredients present, the matrix effects can be considered the same for extracted samples and postextraction spiked samples. Any differences in response can now be considered to be due to extraction recovery.
The matrix effect is determined by comparing peak areas of neat solutions spiked (postextraction) into blank matrix with those of other neat solutions. Because the analytes are not extracted, the analyte should have the same response in postextraction spiked samples and in neat solutions. The matrix ingredients, therefore, cause whatever differences are noted in the responses.
A useful method to assess matrix suppression is postcolumn infusion of an analyte into the MS detector. The extracted blank matrix is injected by an autosampler onto the analytical column. The setup is shown in Figure 3. The purpose of postcolumn infusion with analyte is to raise the background level so that the suppression matrix will show as negative peaks. This setup has been successfully used to identify and troubleshoot matrix suppression peaks.
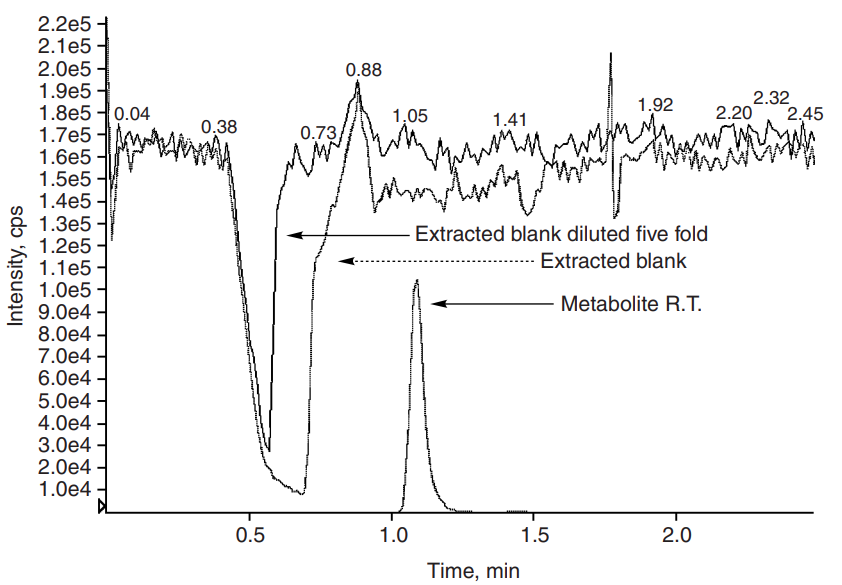
During LC/MS/MS method development for analysis of a nucleoside compound and its metabolite, lower and inconsistent signals of the metabolite peak were observed. An aqueous–organic mobile phase and a silica column were used. The extraction method was a simple protein precipitation. The lower signal was due to matrix suppression, which was confirmed by postcolumn infusion portrayed in Figure 4. A broad suppression band was observed around the analyte peak. (1) The problem was overcome by diluting the extracted sample five-fold with the weaker elution solvent, in this case, acetonitrile. The suppression was no longer observed, and the method has since been validated and successfully used for routine sample analysis.
Reference: Systematic Troubleshooting for LC/MS/MS - Part 2: Large-Scale LC/MS/MS and Automation