What does the successful LC-MS look Like? or What is the principles that produce good successful data? or What do you address when developing a new method?
I want the isotopes to be as they should be.
- whether a molecule is basic, or acidic
So let’s let’s begin with the lecture five, shown on the slide, principles of successful LCMS. The more experience you have in this field, the better you get at developing methods, optimizing the method, generating nice data that’s publishable and presentable, just getting a mess spectrum or my view that looks crappy is not really success, you want to I want the isotopes to be as they should be, I want the chromatographic peaks to not bother are card carrying chromatography chromatography is look at LC peaks and milliseconds, they can tell whether it was a good run, or that the person knows what they’re doing. And showing the data of both the mass spectrometry data and the LC data, or experienced observer can easily tell whether the user knew what they were doing, whether it’s good data or not. If you are in high stakes, environments, like a forensic laboratory, where it goes to court and in my racehorse drug testing days, a $10,000, fine, was like a $5 parking ticket for us, they would gladly pay $10,000. But if they were forbidden to race, in New York State for three months, that’s most of the season that was a big hit. And so they pulled out the top guns, the attorneys, an expert witnesses many a time I’ve gone against expert witnesses, who would pick pick away at the data, try to find something wrong. And if you’re in such a high stakes environment, you want to do good results, you want good solid results, including chain of evidence, but the analytical chemistry itself. And so one of the points of this lecture is to understand the principles that produce good successful data. So developing a method and LCMS method that means that you have a lot of things addressed and some of those things are on here. And knowing organic chemistry, we are primarily talking about organic molecules as opposed to inorganic molecules. And under I asked yesterday, how many of you remember some of and have experience with organic chemistry, knowing the basics, you don’t have to be an expert at it.
 But whether a molecule is basic, or acidic, that helps us decide whether to do positive or negative ion. Simple as that is that heat sensitive, I mentioned classes of compounds are known to be heat sensitive. Typically, they’re peroxides. That’s an oxygen oxygen bond, single bond sulfoxides and oxidize sulfur cell phones is a doubly oxidized sulfur, nitrogen, can they oxidize the oxides, these are weak bonds and are labile sensitive to excessive heat, even the heat that we subject these two, as a brief digression, you know what
But whether a molecule is basic, or acidic, that helps us decide whether to do positive or negative ion. Simple as that is that heat sensitive, I mentioned classes of compounds are known to be heat sensitive. Typically, they’re peroxides. That’s an oxygen oxygen bond, single bond sulfoxides and oxidize sulfur cell phones is a doubly oxidized sulfur, nitrogen, can they oxidize the oxides, these are weak bonds and are labile sensitive to excessive heat, even the heat that we subject these two, as a brief digression, you know what the Leidenfrost effect 1 is,
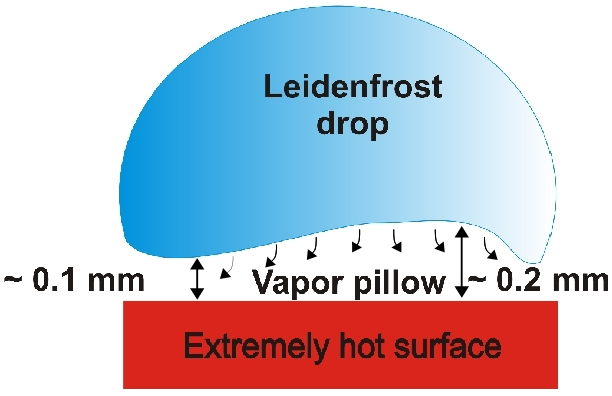 if you ever iron your clothes, and you want to know when the flat iron is hot, enough to iron, one of the things you could do is this and touch it. That monolayer of your saliva liquid water does a steam quick steam distillation and creates a barrier so you don’t burn your finger. If you touch your dry finger to the flat iron, you burn your finger. But if you do what I just suggested, that’s
if you ever iron your clothes, and you want to know when the flat iron is hot, enough to iron, one of the things you could do is this and touch it. That monolayer of your saliva liquid water does a steam quick steam distillation and creates a barrier so you don’t burn your finger. If you touch your dry finger to the flat iron, you burn your finger. But if you do what I just suggested, that’s the Leidenfrost effect. Google it, you’ll see. in LCMS, what that’s what we have, we have our molecules surrounded by water vapor. So in a way, many of these are molecules are stable, because they’ve got a moisture layer around them. And even though it’s hot, they’re protected by a layer of water, but extremely thermal thermally labile molecules like those I just mentioned, are so labile that even that little layer of water burns their finger if you will, or Pyro breaks them down. And so the good news is the Leidenfrost protects our molecules in many cases, but not all.
So heat sensitive is the molecule heat sensitive that should there guide you as to how much heat to use in the capillary. Our inlet capillary the first surface that will the second surface that our molecule sees. The first surface is the sprayer itself, the heated sprayer, if you will, and the second is the heated capillary. And then there are heated surfaces inside the vacuum system but generally our molecules don’t see those surfaces they’re in the gas phase. And if they hit a surface we lose them we never see them. So inside the vacuum system that’s it’s all okay it’s in the heat of capillary primarily is the molecule Seibel in the mobile phase and conditions that you’re using that’s generally case but it’s possible to have precipitation occurs, we are doing evaporation, the droplets remember go from larger to smaller and smaller, if the molecule is partially soluble in that mixture combination, it can actually form a precipitate at a particle We don’t ionize particles, we will have poor sensitivity, I’ve seen people that have actually done are doing that. And without knowing that they have terrible sensitivity, it’s easy to blame the mass spectrometer. But as I said yesterday, if you don’t get the eye into it, it’s not going to show it any results from it. And so what we do in the process is important, sustainability is important, and is the molecule polar or nonpolar polar molecules are nicely done by electrospray. If it’s truly a nonpolar molecule, it may or may not work with electrospray. But a very well may work by APCI. So these these choices that you are should be making as the captain of your own ship, you’re the chemist, you’re the one setting out to develop a method on a new molecule, a new drug or new whatever the situation is, if you’re in a lab, a lot laboratory like Andrea was with an EPA laboratory, there are methods already established, and you’re happy to follow them, the method development has already been done. Oftentimes, it takes five years to get that method developed, and approved, because it’s sent out to round robin laboratories, everybody tries to do it. And they all decide when it’s right. And then everybody must follow that that’s not developing a method that’s using one that somebody else has developed. But in our world, in many people’s world, you have a new compound a new situation, and you have to develop an LCMS method. So that’s developing the choice of the ionization, the choice of the chromatography, the choice of the mass spectrometer, so sometimes, if you need a high resolution, accurate mass measurement, our instruments not going to help you. It’s a quadrupole. It’s a good quadrupole. But it’s not high resolution. For that you need a Fourier transform spectrometer, a time of lightness and mass analyzer, or an orbitrap, high price tickets, half million, $300 million. So choices, the tools that you use, the iron source selection, electrospray, or APCI, we’ve touched upon that, importantly, the column selection, LC columns costs quite a lot from a cheap one is 150 to $200, expensive ones are $1,000, they don’t last forever, a good column will handle a appropriately, will maybe run 1000 samples. So it’s a consumable. And because it cost so much, there are many different columns out there, you probably can’t afford to have an example of all of them, you want to have a representative range of polarities from reverse phase, like C 18, to a medium polarity, like on amino propyl or cyanwood, propyl, two very polar columns, like a silica column or a Hilic column. You don’t want to have all of these. But when you’re developing a method, you don’t want to place an order and wait three days or a week to get a new one, you should have a range of three to five different polarity columns. So you can pull it out of the drawer and put it on your system to see how well it works for this method that you’re developing. It’s true, though, that might many people use only see a teen, you can sort of get away with that, but many of your separations will be suboptimal, they will not be as good as separation is if you use the appropriate column for the compound that you’re looking at because of its polarity. We have normal phase columns that are that are silica without any stationary phase bear silica is what a normal phase is. And by the way gradients, a gradient program is not relevant to normal phase, we often almost always use gradient and reverse phase chromatography, but a gradient in a normal phase is not done, it’s almost always isocratic. So the mobile phase composition you can go from zero water to 100% water or zero methanol to 100% methanol. And it’s usually such a combination those common mixtures are aqueous methanol, or aqueous acetonide trail there, there are subtle differences between them. And it may be your personal preference, or there may be some reason why one is going to be better than other. So reverse phase and notice how I’ve written it, it’s Edie hyphen, if I review your paper, I will be picky and point that out not a big deal that you want to appear as smart as you are. And so these are polar aqueous organic solvents with additives, meaning often acid we acid such as formic acid or acetic acid or if you’re going to have a basic mobile phase, then it would be ammonium hydroxide trimethylamine triethylamine, some of these low molecular weight species and normal phase solvents or nonpolar solvents such as hexane is the most common one. And you have the choice are you going to have a gradient separation or or isocratic separation, a gradient newsy is analogous to a temperature program and GCMS you must have done lots of GC temperature programming. It’s It’s It’s solvent mixture programming in this case, and rather than having a runtime of 45 minutes while like this, if you use a gradient, or a temperature program, you shorten the runtime, you also compress the peaks, you narrow the peaks. If you’re doing quantitation. That’s a good thing because you’re getting the area under the peak that’s the constant durational that’s a big fat broad peaks is, is less precision and accuracy for your software to define the baseline of that peak. And very importantly is the sample matrix complexity.
I mentioned yesterday, we’re I’m just became interested in breath analysis, the matrix in the breath is mainly lipids, not all kinds of proteins and in phospho, lipids and so forth, although I think there are some of those in there too, compared to urine with lots of salts and proteins and biological matrix, biological matrix, these are very complex. And to have good LCMS results, we need to do adequate sample preparation and optimal chromatography, those two things combined give us really good results. Now, you should be aware that there it is possible with certain systems to directly inject plasma, so thermal not dimension competition, but other main main companies acquired a company A while back, that was that produces what they called turbo chromatography. To me, it’s an exaggeration of the facts. But they actually that capability has a combination of software columns, and mass spectrometer, so they directly inject plasma into the LC system. And these are popular in clinical laboratories, hospital laboratories will dollar route all due respect to clinical laboratories. That’s where our blood goes when we have a blood test. These laboratories are run the people there are medical technologists, they’re not often PhD at organic chemists, they’re they’re good capable technicians, but they don’t have the time or they or the knowledge to develop high tech methods. And so they like the idea of directly injecting plasma and getting a result in this system from thermo, I forgot what they actually call the product, but it’s an integrated system of directly injection biological samples to do LCMS quantitation. It doesn’t work for all applications. One of the benefits of a sample extraction
is concentrating the analyte, the target compound in the extract, if you have plasma, you’re not concentrating and you need really good sensitivity to have that work. But the only point of that is that it is actually possible in certain instances to directly inject plasma but most of case in our world that’s generally impractical to do that that’s a special system. The original company was called cohesive technologies. It was formed by scientists EVO that spawn out of waters and develop this and thermal purchase them. And so sample matrix complexity is really important. Directly objection directing, biological sample is quite tough. I have an old slide that I don’t have in this course, from the racehorse drug testing days, my wife would say, why don’t you just connect a mass spectrometer to a horse? Well, horses run 25 or 30 miles an hour, but I have a plastic horse a full size plastic horse that my student stole borrowed from Cornell was on a platform, we backed it up to the mass spectrometer put a piece of pressure tubing from underneath its hind legs to the mass spectrometer, and that’s the horse ms interface that doesn’t work very well. Doesn’t it’s just a joke and it doesn’t work very well. So the property the compounds, is it acidic or basic? Is it heat sensitive? excitability? And is it polar or less polar? These are the points we just discussed in some detail. ion source selection, so you’ve got tools in your toolbox and they are two of them or are you going to do electrospray or APCI. In many cases, either one will work in most cases one will work better than the other. So polar compounds are course are well suited for electrospray. Basic compounds can be very positive. And what I like about a basic compound with its basic it usually means that has a nitrogen in it a basic nitrogen, try meth try alkyl amines are the most basic Amman’s are much less basic, but still have some basicity to other hetero atoms, non carbon atoms that are useful for adding a proton or oxygen. And that’s where this term proton affinity comes in. Such molecules have a certain proton affinity that can be measured. It’s generally not available to us unless we look it up for the molecule. But the affinity of that molecule to a proton determines whether you can ionize it or not whether the ion the proton sits on that molecule, we talked about the PHS the polynuclear aromatic hydrocarbons, they have no hetero atom, and there’s no place to put or accept a proton. And so we formed ions by charge exchange as I explained yesterday, no protonation less polar compounds. I mentioned steroids, lipids, cannabinoids, there’s quite a class of compounds that are considered nonpolar. And these are often better done more successfully by atmospheric pressure chemical ionization. The ion source optimization you experienced that yesterday you’ll learn the three or four parameters on our ion source that every mass spectrometer has related parameters voltages, and And those optimizations help you optimize the performance, mainly the sensitivity, the main thing that’s doing is, is improving the sensitivity. The mass resolution and mass axis calibration is not such as partially sensitivity, but it’s its peak shape and quality of the mass spectral data. And so and source optimization says, See lecture three Part B addicts, this is the the frustrating to novices that want just the molecular ion. And right at the beginning, remember I said that protonate, a molecule is not the molecular ion its terminology. By mass spectrometry definitions, the molecular ion is what you get an electron ionization, it’s m dot plus, it’s the molecule minus one electron. So it’s masses and is basically the same. So these protonated molecules, and as I said before, it’s not correct to say protonated molecule ion, because that’s redundant, of chemists knows the protonate molecule is going to be an ion. And so we have the formation of protonated molecule is generally a very abundant ion. But in electrospray, we see addicts, so here is a 393. And if you do the arithmetic, and it doesn’t get real hard, all you have to do is subtract 361, or 360, their actual molecular weight from this, and you’ll have a difference of 33 atomic mass units. And that’s because methanol bound by a proton is a bit attached to that molecule. So it’s a a protonated methanolic, Iam m plus 33. If you have an M plus 32, plus 23, what’s the 23? Sodium? Where did the sodium come from, you may have put it in there. Or it may be your mobile phase water sitting on the bottle for days and days, and leaching out in fantastic tiny amounts of sodium from the glass, there’s glass of the sodium and potassium in the glass bottles. If you use plastics, some people use polymer of high quality laboratory polymer plastics, so you don’t get sodium in there. It’s not a bad thing, you can consider it an added confirmation like your wait. But what about this guy way out here. I didn’t mention this yesterday. This is where your organic synthesis people that think more is better. What happens this is a proton bound dimer. This is twice the molecular weight of the molecule in this instance, bound by a one proton. So there’s a bridge, there’s a proton in the middle, and the two molecules are here. This is a very reproducible artifact of injecting too much sample, the molecule is so concentrated in the gas phase, that they’re interacting with each other. And so what what happens to a novice and we have lots of customers that are new to mass spectrometry, and they think more is better. And they’re looking for the molecular weight. And let’s talk through this scenario here. This is the molecular protonated molecule that we’re looking for. In here. It’s quite abundant. But if one injects higher and higher concentrations of that molecule, mean, while they’re looking for that molecule, and they’ve already injected, maybe 100 micrograms, or 50 micrograms, way too much material, they run the experiment in electrospray. This applies mainly to electrospray. And they will not see this either at all. And by the way, they’re expecting a 361 ion. And so maybe they scan to 400. It goes to 2000. What are you saving for and what’s happening is this, this has been dramatically reduced, because it’s all go into this and they don’t see this because they’re not scanning out there. So I had my own homemade rule, I call it the hennion to to molecular weight two times I’m like wait plus 50. So you cannot be fooled by something like this on an unknown molecule A new molecule to you. When you when you scan full scan, run at least twice the expected microwave plus a bit more Eisley just 50. And then you won’t miss something like this. And so the typical scenario is they see this very weakly. And they think they have poor sensitivity. What they don’t know is all Ali Mala ions are going up here. So they inject even more in this goes down. And they’re still not scanning out here seeing this. So they’re not knowing what’s going on. And they get really frustrated and call us and what’s what’s going on. So be alert to that, especially when you have students using these things, that proton bound dimer and electrospray is a common artifact of too much sample. And it’s so common that what I just described, they don’t scan up far enough and they see a weak peak. So they injectable more and it goes down that drives them nuts. So be aware of that. In this bottom example, now we have atmospheric pressure come on. Chemical Ionization, by the way, this is this molecule is steroid and like your weight 360. So we saw the 361. And that same molecule under APCI conditions, remember this is a non relatively nonpolar compounds. We see the protonated molecule and sub fragmentation. Now, in this example, we see minimal fragmentation because electrospray is mild ionization some mild puts in parts very little energy to the molecule or to the ion APCI is also very mild on isation. But then when you have, if you’re an organic chemistry, realize that’s a tertiary alcohol, and it’s adjacent to a carbonyl. If you protonate that, that oh, ah, you have the incipient loss of water, it can readily lose water perform an lol cow diet or, or a conjugated CarBody de unsaturated system. So any amount of heat at all, doesn’t take much at all to facilitate the loss of water. And that’s what we have here, that you might even have loss of two water molecules from them. And so ionization is mild, but the heat associated with this process can cause some fragmentation, not a lot nowhere near as much as you get an electron ionization and electron ionization, it’s rare to even see the molecular ion. Because this molecule breaks up in so many ways. The E IMS spectrum is very complex for steroids, many, many fragments.
So that’s enough on that. Moving to chromatography, there are a number of choices. Most of us do not use some of these, some of those that are least used by small molecule people are size exclusion chromatography, this would be used for for proteins and biological applications. There’s ion exchange chromatography, there is normal phase and reverse phase. In the vast majority of applications are reverse phase chromatography. Ion pair chromatography has some role in LCMS applications. Not shown here are two or two relatively new kinds of chromatography, and that’s called Haylock. Hydrophobic interaction liquid chromatography. That’s an interesting new entry that can be useful in LCMS. If you have a sample that’s got a number of very nonpolar molecules in the presence of a novel, it’s a number of other very polar molecules. These these hillock columns can handle both of those fairly well. And another one is hmic. Hydrophobic interaction chromatography. This is relevant to buy biologics, proteins and peptides. And we, I’ve had a fair amount of experience with those, but most people using our instrument, do not use HBIC columns. So the two that are most important to your world are normal phase and reverse phase with emphasis on the reversed phase. Ca, teens cyclopropyl, amino phenol, each of those has varying degrees of polarity, not ca 10 being the most nonpolar columns, then available. The columns, of course look like this. Within the column is where the separation occurs, the proper choice of column is critical or very important. You want a nice chromatogram. If you have lots of samples, you don’t want 30 minute runs. If you do the arithmetic, you’ve got 100 samples, how many hours does it take to run all those samples? If you don’t have large numbers, I get the impression. None of you have high volume, sample volume throughput, you’re not running hundreds of samples. So that’s less relevant there. That the types of columns that you have an HPLC there are analytical ones with inside diameter. If you tell me about your method, and you are asking questions, one of the first questions I’ll ask is what is the inside diameter of the column you’re using? Because that because that dictates the flow rate. And remember the firehose effect we don’t want the mass spectrometer feeling like it’s drinking from a firehose. The most common inside diameters for most people today is 2.1 millimeter inside diameter, and that those optimal work optimally the optimum linear Vaci is about 200 microliters per minute, instead of a milliliter per minute. Historically, the one mil per minute columns are the 4.6 millimeter inside diameter, as long as 15 to 25 centimeters long. Those are almost dinosaurs. For most of us, most of us use much shorter columns, five centimeters long, 10 centimeters long being a relatively long one. preoperative columns have larger inside diameters because you’re loading a lot more sample on the that’s that’s a factor to or an important fact that the insert in addition to the floor rate, the inside diameter determines the amount of sample you can load on the column. If you were to take one extreme, I guess I don’t have here the Nano you’ll the nanoscale column is less point one millimeter is 100 microns. And so nano columns are 75 microns to about 200 micron inside diameter. The sample capacity that column not surprising is very, very low. You cannot inject 50 mil nanograms on to such a column. It’s usually sub nanogram. So if you want to inject large amounts of sample on the column, you need a larger inside diameter. That’s what preoperative columns are where you can collect milligram quantities and sample instead of nanogram amounts. What are the columns made of most of them are stainless steel, they operate under high pressure. They in the old days, 6000 psi was considered really high. Now we have UHPLC or ultra high pressure ultra high performance ultra high price I like to say these are not not cheap. They have 1.7 micron particles, various tiny particles. And they operate under very high pressure as much as 25,000 psi, typically 10 to 15,000 is fairly normal right now we have a problem in the lab right now with one of the injectors from one of the vendors, and it’s leaking, and when you get above about 5000 psi, by the way peak tubing is, I’m not sure I get to this later, but I want to make sure I cover this. The peak tubing is a wonderful thing compared to stainless steel tubing, you don’t really need wrenches, it’s easy to cut and handle we used to be all stainless steel, but peak tubing has a pressure limit. Do you know what the pressure is? What’s the maximum pressure if you have peak tubing connecting your column to your injector and to your mass spectrometer and what what’s the maximum pressure P can
handle 3000 10,000 4000 absolute maximum 5000 You’re safe at 4000 psi. So if you go to UHPLC you should have noticed that you don’t use peak tubing in that it’s now all stainless steel fitting tubing with special fittings. That’s another thing you have to have. You need a little chest of drawers in your laboratory where there are certain ferals and fittings for for certain pressure regions. If you have UHPLC. It’s a separate set of fittings and ferals don’t mix those two high pressure names high headache if you use the wrong fittings because they will leak in leaking is you know, you can’t really tolerate leaking. So they’re also glass. This is glass columns. But that’s really low pressure, of course, for mostly for biomolecules, and right quite uncommon. But peak polymers are also common. And that’s made it easy to make fittings and so forth. But there are pressure limits to it. It’s also true that peak is somewhat softened by a CDI trail, you can use a CDI trail with peak tubing, but it will not last as long it softens in animals the leak in a few weeks, if you will. What is inside the column I hope everyone knows that or has thought about that sometimes particles, silica particles finally sieved, hopefully a narrow distribution in size, you don’t want them ranging from 1.7 microns to 10. microns, you want them ideally to be monodisperse or model the same all the same size. The puck part was small diameter, sometimes porous particles, that means this hole through them, there’s a lot of information and more than you need to know here. And I won’t go into too much detail. But there’s a lot of science in modern developments in packings of columns. The most popular comment sizes are up of particles are five micron, that’s actually less popular these days, they used to be common, then they went to 3.5, or three microns. And now they’re 1.8 or 1.7. By that number, I can even tell what company makes it because waters makes 1.7 Somebody else makes 1.8. They’re all less than two microns. And that’s very, very small particle, they’re packed using high pressure there, they should, you should not really take them apart. If you disturb the bed, if you lose any of the packing, you’ve got a void in the column. And then that causes extra column variants that your peak shapes will start to be broadened and so forth. So you’ll want to treat them very carefully. Porous particles means that some of the solvent could go through the particle a kind of a tortuous path, that’s a good thing because it gives more time for the exchange of the molecules to the surface to take place. The stationary phase is an interaction between a competition between your analyte that’s in solution going through the column, if there was no packing, it would go straight through and not be retained, we want to have relative retention. If we have three peaks separated, they all are interacting with the surface the stationary phase in a different way to a different different extent, the one that comes out first the peak comes out first was the least retained, they did not have much interaction with the stationary phase, the peak that comes out last is the most retained in those cases. And the idea is to have them separated in that column and sent one at a time to the mass spectrometer. So the process of retention of the sample components, the analytes is determined by the choice of the column packing. In most cases, the silica is pretty much the same. People will sometimes buy a column from vendor one, it’s a C 18. And all the specs look the same to buy the same column from vendor two and it behaves differently and they will the vendors have their own tricks they do certain key things and so I see a T does not see it and see it but how the vendor has prepared the column may give you different results. They may be subtle differences or there may be major differences. So don’t assume that all ca teens are all mamita purples are the same
So here’s an LCMS system, simple situation or even whether it could be a UV detector, and we zoom in and see that there are particles, they are often spherical. Sometimes they’re not spherical, you will need to get in all those details with a tightly packed column with no significant voids, if you will, and the column inside diameters are can range from what they used to be 4.6 millimeter, they still are available, and they the optimal mobile phase flow rate is 1,000,001 milliliter per minute. The ones we prefer in favor of the 2.1 millimeter ID, why are these fractions that just the stainless steel is not made by the vendor that makes the columns they buy the stainless steel tubing from from some company, and that happens to be what the inside diameter is, it’s easy to make a 2.1 millimeter ID. They’re also a 1.1 millimeter ID there are 300 microns or often 320 micron IDs is moving towards capillary columns, if you will. And finally, the nanoscale columns at 75 microns, operating it to use the 200 micro nano liters Well, while 100 to 200 Nano liters per minute, which is 1000 times less than 200 microliters per minute. All these are amenable to LCMS. But the vast majority of folks that don’t do in conventional LCMS, use 2.1 millimeter. As I mentioned yesterday, the only reason the driving reason to use a smaller inside diameter is under electrospray conditions where you want more sensitivity. Here’s a question for you. Is there any benefit using a PCI if you if you’re looking at steroids and a PCI works well for you? Can you gain sensitivity by going to smaller and smaller ID columns? No, that doesn’t buy you a thing normals no one, there’s no justifiable reason for using smallbore columns for a PCI 2.1 Thank you very much works just fine. Some people will even use 4.6 million at millimeter millimeters ID at a mil per minute. So a PCI does not benefit from nanoscale chromatography, the length of the column, the longer it is up to 25 centimeters, the more theoretical plates more separating power, but the back pressure will be higher, and the runtime will be longer. Short columns three to five centimeters give fewer plates, lower back pressure and shorter run times. So it depends on what you’re trying to do next door with our CRO with lots and lots of samples we want short runtimes even less than a minute if possible. So we generally use very short columns as in to two centimeters to three centimeters something like that, because you cycle time for per sample a per plate is going to be much less mobile phases. The mass spectrometer does not like drinking from a firehose, the preferred flow rate is point two to a half mil per minute, that’s 200 microliters to 500 microliters per minute. The flow rate is usually dictated by the column inside diameter. As I said 2.1 millimeter ID columns are preferred. And that works very nicely in this range. The longer the column the better the separation, but the longer the runtime obviously. mobile phase solvent, they also the solid in the in the column is called a mobile phase. And that term determines what the spray solvent is a coarse solvent mixture containing water and methanol or st nitrile. The higher the percentage organic solvent, the better will be the I’m sorry, the higher the percentage the organic solvent the better but this will reduce the separation. Why methanol is a strong solvent and reverse phase it really eludes the sample the analyte quickly where water is a weak solvent, it has the less ability to elute sample off the stationary phase. And so it’s a compromise. And we often start with high aqueous so that molecules can be trapped on the column and head gift have them have time to separate as they go through the column. And we increase the methanol percentage through that gradient to induce them to eluded the other end of the exit of the column. Reverse phase columns and mobile phases, nonpolar columns and polar solvents. as I just indicated, we start with high aqueous and go to high organic and this is appropriate for those compounds that are quite nonpolar. They’re going to interact a nonpolar molecule like dissolves like ca tannins really nonpolar think of a wax. And that’s what your stationary phase is. So very nonpolar molecules will will linger reside in that longer than polar ones. It’ll go right through normal phase columns with just silica
or use nonpolar solvents like hexane, hexane ethyl acetate, chloroform, usually it’s a very high percentage of hexane 98 97% With just one or two or 3% of the one of these other ones. gradient Lucian is not applicable not use a normal phase LC. And the compounds are typically very polar. For those for those applications do Ain’t Heavy ever, any of you ever use the normal phase column for LCMS hexane, for example, let me make a caveat. That’s really not any more something to worry about. But if you’re using APCI, and if you’re using normal phase chromatography with a 98%, hexane, and if you have a discharge needle with a spark on the end of it, should you be worried about hexane exploding. hexane is very inflammable. By the way, you’re using nitrogen, which keeps out oxygen, you need oxygen to have an explosion, and you’ve got an enclosed plenum housing for your source. So air from the room should not be getting in there. So it really is not a problem. But it could be if you were letting air in, or if you’re using air as a nebulizing gas. So you do have hexane think fire in the presence of a discharge needle, think needle mean think these are none of this is rocket science, but it means being aware of what you’re doing. So you can do it these modern instruments can do it but be be aware that potential solvents, I think I showed this additives this sequence yesterday, list of common things, call them LCMS, friendly buffers and additives, common solvents, and things, no no’s things not to use, alkali metal phosphates, borates, citrates, inorganic acids, these are things not to use, you can use these or do if you’re doing HPLC UV, the UV detector just shines a light through a cell, you’re not doing mass spectrometry Oh, column selection, think I’ve touched upon each of the points I’ve made here. If the sample is chemically pure, a shorter column can be preferred one to five centimeters long. So I’ve covered each of those points, I think previously isocratic versus gradient isocratic, the mobile phase comp solvent composition remains the same, you start out with 5050 methanol water, and you don’t change it through the whole run. And that’s shown here in this graph with a constant percentage of methanol, for example, a gradient I mentioned earlier is analogous to temperature programming and GC, where you’re starting with a low percentage, usually of the strong solid, methanol might be only 10% methanol, but by the end of the gradient, that’s up to 90% methanol. And then at the end of that gradient, there’s a duty cycle time where you need to recycle the gradient before the next injection, you got the mass spectrometer sitting there waiting for the next injections. And you need to recycle, usually that can be done within a minute or two. But if you’re running 1000s of samples, that adds up. But it’s important to know you need to recycle the gradient, let it equilibrate, and then that’s when you make the next injection isocratic or gradient turns very important factors and parameters and LCMS. Why our mobile phase gradients use an HPLC a variety of reasons. Here’s an isocratic elution. And here’s a peek coming out in 70 minutes an hour and 10 minutes. If you have lots of samples or you’re impatient, that’s a long time to wait nice looking piece nice and sharp, but something is highly retained. Something is much more retained the nice species, eyes under isocratic conditions of 70% water and 30% of the scenic trail. If you do a gradient from 20 to 60%, that peak comes out now in 28 minutes and almost a third of the time. And both by the way, it’s gone from being broad to sharp, if you’re narrow, if you’re doing quantitative analysis, that’s a good thing. If you are doing electrospray it’s a good thing because it’s behaves as a concentration sensitive detector, and the peak volume and that peak being narrow is is gives you a higher concentrations. So there’s two good three good things shorter runtimes narrow peaks are good sensitivity. And what I forgot what the third one was, but there’s at least two good things for doing a gradient like this and saving time basically. And you need a binary gradient system, you need to something more than infusion pump, you need a good pump that can do this. Reverse phase chromatography sometimes called RPC, the column packings nonpolar. CA, CA teen are common examples. Most popular mode, most people use it for everything they do whether it’s the correct or optimal thing or not. They just used to do it they maybe some people have never changed the column. I can’t believe that but they they don’t change them too often.
Any other key points here I think we’ve covered each of these, the feature and one of the other courses I teach I show two chromatograms and ask which one is gradient in which one is isocratic? This is a gradient of course And what it does is give all the peaks are generally narrow if it’s an isocratic Run, maybe if I go back to that one yeah, this is a good example, notice how narrow and sharp these are, the lady looting one is broader. That’s a key chromatography nose can tell without being told that that’s an isocratic Run, because the last peak that comes out are the later pigs come out, they’re all broader, because you’re not compressing the peak by adding in more of the stronger solvent methanol. So that’s typical of a of a isocratic run. normal phase chromatography, it used to be called adsorption chromatography, because there’s no stationary phase is the interaction of the molecule with bear silica, silica think silanol think Si O H, and so ca molecules interacting with that. And so here’s a run in seconds, this is 54 seconds, so it’s less than a minute. For these molecules being separated. Pollen column packing is polar, like silica, a normal phase separations performed in I wouldn’t say this is generally true. They’re not necessarily faster, shorter runtimes than others. The technique is useful, useful for for. For these aspects. You might ask why, or when should I use normal phase, if the molecule is water sensitive? That doesn’t happen too often in my world, but some people have water sensitive compounds. And if that’s the case, then you would not want to be doing a reverse phase chromatography, which always has water in it. If you have geometric isomers. That’s a weak argument in my view, because reverse phase can do that to sis trans isomers class separations, chiral columns, so chiral means you can separate enantiomer as mirror images of each other. I mentioned yesterday, I think that mass spectrometry is a chiral. And mass spectrum of two antibodies will be absolutely identical. Its mass spectrometers are great, but they can’t do everything. So we need that chromatography to separate those and send them one at a time. And so some of the good examples of chiral separations are done with normal phase chromatography. A good modern example is SFC Ms. Remember SS co2 is analogous to hexane and polarity. And so chiral separations by SFC are quite common. They’re often shorter run times faster, usually about a factor of three faster than normal phase. So those are justified justifications for doing normal phase chromatography. matrix effects is the big elephant in the room. I wish we didn’t have this problem, I have a dream of someday, there being a better ionization technique than electrospray, some electrospray urn Don Fen sharing the Nobel Prize, it’s a great technique has revolutionized LCMS. But I have what I like to call happy discontent. I’m happy with all the things that can do. But it irks me that I suffer so much for matrix effects, it causes lots of problems, I’d like to have a ionization technique that was not bothered by a matrix effects. UV is not bothered by you matrix X, Ei, electron ionization is not affected by matrix effects. If you can do a good separation, you can have normalized response. So matrix effects we have to deal with. That means co evolution of extraneous or endogenous compounds with your analyte, which suppress or interfere or sometimes enhance the signal most of time is the suppression of the signal of your molecule that kills us in quantitative analysis, we get a weaker peak than it really should be. And that’s an issue we have to deal with. And the best thing we can do is optimal sample prep and optimal chromatography. dilution of the sample is also advantageous. The opposite device that we talked about yesterday, and you saw briefly, a member of the solution to pollution is dilution. That is a significant dilution effect. And as long as it does not dilute it so much that we can no longer see it. That is the downside. If you over dilute, it’ll be so weak, you can’t even see it. But But dilution does minimize matrix suppression. And APCI. With its strengths and weaknesses is less susceptible. I cannot say that there’s no suppression, but it’s dramatically less than electrospray. So what’s really happening? Why does suppression take place? The endogenous molecule, the interferon is competing for protons with your molecule. And often there’s so much more of it, it wins out. And so your little molecule doesn’t get its share. I’m oversimplifying, but it does not get its share of protons. So we see less signal less ion current. That’s the why of matrix suppression.
chromatography summary, choose an appropriate select selective stationary phase that favors the analyte and what I mean by selective it resolves them from interferences. chromate the stationary phase, it’s called alpha, alpha is the selectivity. A stationary phase gives you different selectivity. That’s it’s a proper correction. term some people misuse the term of selectivity. I mentioned yesterday on early postdoc that came to me with as a card carrier and PhD in chromatography. And he’s the one that changed five columns in a day where I would screw up all day long trying to change gradients with the same column and not achieve anything. And he could move peaks around by changing the column, enhance the stationary phase. So when you’re not getting anywhere, try a different column, and have a few not 100 of them on him that are not another ca teen what another stationary phase a little more polar, a little less polar. The optimum ID is affects affects the sensitivity and the flow rates, the LC conditions to serve the requirements of the project, that’s part of the method development. We met, we develop methods and then we often validate them. And you have, there’s a lot to that, that I won’t go into. But you’re probably familiar with some of those aspects, ensure the mobile phase and conditions are not detrimental to the ionization mode. This is a marriage of two techniques. Remember the bird and fish it was not meant to be, but it can be. And if you’re married for very long, and I want to ask you how long you’ve been married or not married, you have to know what compromises means you have to suck it up sometimes. And at the HPLC has to suck it up and not use citrates. And the mass spectrometer has to suck it up and not take off two mils per minute all the time. It’s a marriage, that’s a compromise and yet we can make it work. You would like to have sharp, symmetrical and narrow peaks. They’re not noisy. We have a term. You know what a chick up LCP, it looks like a chicken head. You know, chicken has a comb A rooster, we don’t want peaks, I was bothered yesterday with with Li Fei pig, I would have done that. Again, I don’t like those bumps at the top of the peak, it should go up and come down like that. There’s reasons for it to happen. I think he had too much sample in that in that case. And so nice, smooth peaks without chicken heads. chromatography is a UV, there’s no such thing as noisy peaks and the early days of LCMS. That things didn’t work. So well. I think I mentioned we used to spray and pray that would work. And so it was often noisy peaks and chromatography. So I don’t want that technique. It’s comi peaks. While we now can do good peaks, if we know how to do it, right? Minimize a sample to sample runtime to achieve the sample throughput. If you have lots of samples, consider helix columns have any have ever used a healer column, these are relatively new. And they are good for handling upsample that has some very polar molecules and some in in the presence of flurry nonpolar molecules. And I won’t go into the details of that. But it’s it could be worth if you’re having trouble with polar compounds that go right straight through your column and retained, then with CA teen, then you ought to try hillock it’ll work much better.
These This is a article along 10 years ago and LC GC and maybe not a peer reviewed journal, but a magazine. So these are column myths that people generate their, their own ideas, one of the things that amuses me or, and concerns me is we have academic incest, and you must see it in in the laboratory like you have or are in commercial laboratories. I bet you saw it some so someone comes in. And maybe they’re an expert. They’re the were the company in the beginning. And they’re the LCMS expert. And they get second to renewing routine samples. So the company hires a new person. So the expert teaches that new person a technician how to do it. And then expert floats to the top and comes a manager or leaves. And now a year later, another new employee comes in and this this, this technician teaches the next person all the bad habit he or she has developed and the knowledge transfer just gets diluted and incorrect. And so there’s a lot of the myths sometimes about what I what happens or how to do it right in the laboratory. And sometimes I wonder how they see Paul. But in any case, these are myths about HPLC. That is that you cannot reverse an LC column in the early days. columns that come along the way as columns came commercial columns developed in the mid 70s. So we’ve been making and better and better columns for years and an early days if you did reverse one other words, you connected the what was the accident as the inlet and it would disturb the bed and be a crummy column. But these columns today’s are very robust and it’s perfectly okay to reverse the columns as you connect them. This one is another one I addressed earlier that all CTN columns are the same they are not vendors do different things little tricks the way they do things, you will get different results maybe suddenly different but not identical results. Guard columns do not affect the separation. The guard comm is an insurance policy to protect and extend the lawn the length the life of a new column. They are usually about a centimeter long, they are a column they are chromatography and they do affect the separation and a minimally they also protect the column from really dirty stuff or party to kilohertz and so forth. So it’s a good insurance policy. Most people like to use a guard column. And in many instances, high temperature always leads to better separations, high temperatures of what GCMs. We have the column in an oven, do you have your column in an oven with HPLC? We do. But we don’t heat it to 350 degrees, the maximum was maybe 60 degrees, why would you put a calm in an oven? One, if you haven’t an air conditioned air conditioning and temperature, the room goes up and down, the retention times will drift you want the column to stay at the same temperature. So a common thing is to heat the column to maybe 40 degrees C, maybe 35, something above your room temperature, but keep it at that. So there are little devices caters that you can put the column in. The other thing is temperature does if you go to maybe 50 or 60 degrees, no higher than 60, the viscosity of the liquid reduces on a pixel sharpen up it looks like better chromatography, it’s not really better, but it’s because the solvent is viscosity is changing. And so the best reason, justification for heating the column is to have reproducible retention times. And that is an analytical benefit. That’s how GC fid that’s how you cold identify something and check the analytical standard has 2.5 minute retention time. And that’s the same as your unknown. So hence are the same. So it does not necessarily need the better separations. See 18 columns provide better separation and CA Not necessarily depends upon the molecule that you’re looking at CA is a more polar column data C 18. Not dramatically sold, but noticeably, so
smaller particles and ultra high pressure always leads to better separations. Yes, if you do it right, you need to be a nano plumber, you need to have connections that are made correctly. You have seat the Sarah Farrell on the tubing. If the extension beyond the feral is too long, it’ll leak because your ferrets Pharaoh won’t see the using the correct Farrells is important as I said before, so if you have the if you have extra column variance is dead volume after the separation after the column on its way to the mass spectrometer having an umbilical cord this long between the column and the mass spectrometer is not good. Because you’re not you’re starting to lose your chromatography and by the way, the inside diameter of that connecting tube is very, very important because if it’s 250 microns instead of 100 microns is a huge viam you absolutely will have broadened peaks. If you have a long connecting tube to the mass spectrometer We usually keep them as short as possible after the column because no more chromatography takes place them. So if you do it the plumbing correctly, you do get better separations UHPLC columns plug more easily than conventional LC columns. Well, that’s not true either. It’s a matter of the frets and how dirty your samples are. If you can plug this column you can plug a regular column plugging meaning there are particulates in your injected sample, hopefully not in your mobile phase. Have you ever noticed as a fret at the inlet tube in the bottom of the bottle that your mobile phases are all that is to minimize or eliminate particles from going to your system, silica gel base packings can be used only for pH two to seven. It used to be that if you had a pH of your mobile phase outside this range, you would destroy your column, they’re much more robust. Now it’s still a good idea not to have a pH of 14, or a pH of one on your mobile phase as you will eventually hydrolyzed or decompose your column. Paying attention to pH of the mobile phase is important but not as critical as it used to be modern columns should withstand at least 1000 injections. Yes, they can. But if you have really dirty samples, like some people can do, they will last for 20 or 30 injections, you can kill a column very easily with a few injections if the samples are really dirty. And an extra large volumes of samples added columns should always be kept tightly to prevent packing damage from contact with the atmosphere that used to be true. They’re much more robust. Now it’s a good idea to have these little plastic nuts or fittings on your cap them but it’s not necessarily necessary anymore. They will be good next week next month. I don’t know about two years from now. But the comments were much more persnickety in the past than they used to be. This was 10 years ago in this article, and I think it’s gotten even better since then. Wrapping things up, here’s the 16 PHS that you wouldn’t think you could do either AP psi or electrospray. If you take this kit that we have that you have, and look at the conditions we’re using water to see nitrile it’s 800 microliters per minute almost a milliliter per minute. We’re using a 4.6 millimeter column was 1.8 micron so it’s a UHPLC column. The gradient as shown here, we’re starting out zero time at 60% be mostly water going to 100 sent, and that stays until eight and a half minutes and then we recycle. It’s this time, a mile a minute and a half is used to recycle the gradient back to where it started. That’s an important step in a great edge and always need to recycle the gradient. The source is APCI. The consorts conditions are shown here. And under those conditions, you get this result. In about eight minutes, you get these peaks of all these 16 of these benzo these PAH is that we used to think you couldn’t do by APCI. It’s still a pretty impressive they can do this. The folks in the petrochemical industry are quite interested in these conditions. Another set of very different compounds cannabinoids. There are 11 of them, I think here that we are working with that are quite becoming quite important, quite popular. These are very nonpolar compounds, as I indicated before, they’re very sticky. I mentioned carryover yesterday. These are
strong candidates for carryover, we have to be very careful admit from injection to injection. Here’s a list and 11 of them, their names, their masses, and so forth, and the fragments and so forth. And here is a chromatogram than selected ion monitoring in less than seven minutes for each of those Ben knee or postdoc who is now an employee has got this mixture down to less than three minutes by optimizing the the method development and using really good chromatography. And for those who run lots and lots of samples, a three minute run is a lot more desirable than a eight or 10 minute run. That brings us in to the end with the references Darnit I took more time than I intended to but those lessons those points in that lecture for those of you who are not just using somebody else’s already developed method, those are important things to questions to ask important points to be aware of as you go forward doing LCMS these references are relevant to some of the things I’ve covered.