Video: 04 Maintenance and Troubleshooting
Hello, and welcome to LC MS, MS 101, part four, maintenance and troubleshooting, where you can come to learn tips and tricks to keep your systems in tip top shape. I’m Crystal Holt with ScieX, and I’m your moderator for today. Today I’m joined with Dr. Karl Oetjen. Dr. Karl Oetjen is a senior scientist driving food, environmental, forensics, clinical and cannabis applications at Sciex. Before joining Sciex he completed his PhD at Colorado School of Mines, where his research focused on non targeted characterization of complex surfactant mixtures, including aqueous film forming foams, which led to the discovery of several per and polyfluorinated alkyl substances that have since been found in a variety of environmental samples and industrial chemicals. Since joining Sciex, Karl has worked with numerous labs creating both quantitative and qualitative methodologies. And now I’ll turn it over to Karl.
Thanks, Crystal. And thanks, everybody, for joining us for the last session of the LCMS 101 series. And we’re gonna be talking about maintenance and troubleshooting. So something that’s really important, I’m sure all of us at this point have gotten to the lab and had an issue. And whether that issue meant downtime, or some corrective active action needed to take place. It can be frustrating. But there are a lot of tips and tricks we can do to kind of prevent these issues, which is the best case scenario, right? planned downtime is a lot easier to work around and unplanned downtime. And then inevitably, when these things happen, some things to kind of troubleshoot quickly and some just some tips to to get you there a little bit faster. So this will be broken up into three parts. First, we’ll be talking about some preventative maintenance and some regular maintenance that you can do on your mass spec and LLC. And then we’ll go through some data troubleshooting. So kind of a fun case study experiment where we look at some of the more common things we see here at Zach’s when it comes to issues.
Mass Spec Maintenance #
Slide #
[2:28] But first is kind of dive into this mass spec maintenance. So I took a second to think about what are some of the key things I think of when I think of preventative maintenance when I think of keeping my instrument up and running the things that we do in the lab here.
And the first thing is, we always use our diverter valve. That might seem pretty simple, but it’s really important. So we talked about this during the fundamental session a little bit and certainly during the method development session, but that Diverter Valve are essentially putting towards waste when we’re not seeing our analytes of interest. So when our analysts of interests aren’t eluding, we can go ahead and have our flow go right to the waist. So typically, what that would look like is in the beginning of your method, let’s say your first peak Lutz at around three minutes, up until maybe 2.8 minutes, you’d have all your flow just go into your waist. So nothing has been introduced to your mass spec, which means all those salts, all those things that didn’t get retained by your analytical column, are just going into the trash essentially, the same thing you can do for the end of your gradient. So during that whole period, where you’re holding an organic after your last peak Lutz once your retention window or your MRM window has passed, you can go ahead switch that out the waist, and just like all of those more hydrophobic things that are sticking around on your column longer. Again, straight to your waist, never introduced your system. So just preventing your system from getting dirty, quicker.
The next thing that you can do that I always recommend doing, especially if you’re running a little bit dirty your samples. So maybe if you’re running something like a serum or you’ve done even if you’ve done you know a traditional protein crush, looking at a soil sample with some cleanup, just giving your instrument a quick wipe down daily is a great way to keep your instrument up and running longer. And this really shouldn’t take very long. You come into the lab, you take off the source and you can take off that curtain play all without venting your instrument at all. So just connect your hardware profile, take off that curtain plate and give it a quick wipe down with the kimberlite. So take a chem wipe with some water you can add a little bit of acetic acid and some pseudo nitro and wipe off any sort of particulates that have built up on your curtain plate. So if you’re running a really nasty sample, I I always go back to cannabis because that’s that’s one of the the nastier things I have to deal with. You’ll notice some some brown built up, we just want to eliminate that. So we ensure our items are getting into your mass spec the way we intended them to, again, shouldn’t take more than two or three minutes.
The next thing you can also do is check that probe. So when it comes from our LC to our mass spec, we want to ensure that flow is proper, and we don’t have any clogs. So an easy way to do this. It’s just to allow our LC to flow to a mass spec with a mass spec, essentially in standby, and just watch for drips, we should see a constant drip out of that probe. We shouldn’t see a pressurized spray, if your instruments in standby right. So we’ll just see concentrating. And that tells us there’s no clogs. If we do see a pressurized spray, that’s a big hint that, hey, there’s likely a clog in our probe, we can try to clean it, you can take it out and sonicated and methanol, for example, what are they nothing at all. Or you can just replace that electrode altogether, which is probably the best practice.
And last but not least, our roughing pump. So this is something that I added in, based on a personal story of mine were I joined a lab and there was an old GCMs single quad in the corner. And I don’t think anyone had used that instrument in maybe five or six years since I got there. And I certainly hadn’t touched it. And some time went by none of us had really paid attention to it, it seemed to be fine. And then we noticed there was quite a big oil spill underneath that aspect. And what had happened is over time, maybe years, the oil had drained out of that roughing pump and caused a lot of damage to that roughing pump, and we ended up having to replace it. So is that a silly accident where we didn’t check the oil on that roughing pump, but it’s something that can pass on to you guys is make sure that you’re taking a look ensuring that there’s enough oil and you’re roughing pump, your instrument is going to be able to stay up longer. And you don’t have to worry about maybe replacing a roughing pump or even worse some damage to your mass spectrometer. So something super easy to do every month or so just give a quick peek in that cabinet, it can be easy to forget that it’s still there, you’re roughing pump that is and make sure that oil is at an appropriate level.
LC System and Column #
Slide #
[7:25] Next, we talk about the LLC. Let’s move to what can we do there just to make sure that we’re up and running as much as possible. So when we think about maintenance, from an LLC standpoint, one of the big things that I like to sort of bring up is to ensure that we’re changing our mobile bottles, mobile phase bottles frequently, it’s less important for the organic. But I’ve been to several labs where the mobile phase A or the aqueous looks more like a swimming pool that hasn’t been cleaned in a while then through mobile phase. So algae will grow in these bottles. And over time, you can end up with this pretty nasty film that can be pretty difficult to remove from your TV. Honestly, it’s just easier to replace the tubing. So when you’re going into your LC system, make sure that you’re not just refilling the bottle, it’s okay to do for sure, if you’re just in between runs. But we don’t want the same bottle on top of your LC for weeks and weeks and weeks, you’re just inviting sort of the growth of some algae, that’s going to be a pain for you to get rid of. And the next thing that I always kind of recommend doing is going ahead and purging your instrument when you get in the morning. When you’re building your batch, it’s an easy thing to do. And this just ensures that there are no bubbles that have been sort of left behind when it was sitting overnight. And only takes a few minutes and just gives you the best data quality you can. And finally, the last thing from sort of an maintenance or preventative maintenance standpoint that you can do is every once in awhile, go ahead and switch your Mobile phase a line and your mobile phase b line. So put your mobile phase a line in the organic and you’ll be in the aqueous and just purge your system. And the reason to do this is there can be built up the salts etc. in your pumps, and water usually does a much better job of getting rid of these salts than organic. So purchase system and then go ahead and don’t forget to put it back. I’ve done that your data will look quite bizarre if you forget to just switch but switch them back and then purge again. So just the easy thing to do every few months to ensure there’s no buildup of salts in your pump. So another thing we can check out is our mobile phases themselves. So this is actually from some data from a project that myself and a colleague are working on and the group is looking at solvent purity They’re doing some semiconductor analysis. And essentially, they need the solvents to be as pure as possible. So they asked us to dry down a few solvents, and see what impurities we could find. So here in the pink, I have an HPLC grade versus in the blue, I have an CMS grid. And what you’ll probably immediately notice is there’s a lot of things in that HPLC grade. Grated, this is blown down, so it’s concentrated. But still, all of these components analytes features would make their way into our mass spectrometer, and essentially, over time, dirtied up our mass spectrometer. So we want to eliminate them as much as possible. So using high grade solvents is an easy way to do that. Another thing is to make sure that we’re using high grade modifiers as well. So when it comes to our formic acid, ammonium acetate, etc, that we’re using LCMS grade modifiers, just to ensure that our system stays as clean as possible. If we think about how we inject dirty samples, right? Well, we’re only injecting a few microliters most of the time compared to our mobile phase, which might be at 0.6 mils per minute for 10 minutes, for hours. So if there’s a ton of things in our mobile phase, that’s going to really contaminate our system pretty fast. So let’s try to avoid it. And that will just end up with more uptime.
Slide #
[11:23] So, from an LSP perspective, one thing I can’t stress enough that I’ve been guilty of is make sure that you do regular maintenance. schedule it. Personally, I find it easiest to do once a year when you do your mass spec PM. So when your engineer comes out, or if you do your PM, your maintenance yourself, do your LC maintenance as well. These instruments are operating at high pressures, they’re moving constantly, we have pistons, we have rotors, we have a lot of moving parts that just get worn up over time and need to be replaced. So it’s an easy thing to overlook. But it’s really important thing to keep in the back of your mind and to kind of schedule for. So in my opinion, doing it once a year is pretty easy to schedule for it because you just do it at the same time to do your maths back again.
And then something to do when you kind of get into the lab, something to notice that I’ve been guilty of is checking for crimped lines, especially if you have a column of and where the line sits sort of outside the column of and a little bit. It can be really easy to shut your peak line in that column oven and you end up with a level crimp. And you’ll kind of see an increase in pressure. So anywhere that you’re constantly or frequently. Connecting, disconnecting tubing is a good place just to inspect before you run your first sample and say hey, did I accidentally shut my line in my column oven, for example.
And finally, we’ll be talking about how to use this. But having a system suitability test is really important. And this can vary depending on the type of lab you are really all assessment suitability test is, is a known method where you can compare current results to previous results. So let’s say your mass spec has a lifespan of 10 years, over those 10 years, it’s a method that you’ve run frequently. So you can say, hey, you know, over the past month, I’ve lost a quarter my sensitivity, etc. So this could be just the analysis you’re doing or the essay you’re doing. Or in our case, here at Saks, we just use the pharmaceutical mix, that’s very simple and stable. Can we go ahead and check that periodically? About once a month, and just keep an eye on our instrument to make sure if we do notice those drops in sensitivity, we can plan for some downtime.
Slide #
[13:43] And last but not least, when it comes to maintenance, there’s also some computer maintenance. Without our computer, we can’t run our LC, we can’t run our mass spec, we can’t analyze our data. It’s a really key portion of the system. So just a few tips for making sure that you kind of eliminate as much downtime as possible. Number one, well, first of all, make sure you’re backing up your data, I can’t stress that enough. The best place to backup your data would be some sort of network drive, or somewhere electronically. So in the cloud, for example, you can use a hard drive, but just remember that hard drives can fail. So if that’s the route you want to go, having a backup of your backup is important. You worked really hard for that data. And you want to make sure that you have access to it when you might need it in the future. The next thing you can do in terms of preventive maintenance is keep those file sizes down. Speaking from a science perspective, when we saved data, we saved them in a data file, that’s called a wiff file within that wiff file are all the samples. So you can think of it almost like as a batch. So within a single file, there might be 30 110 samples. But what you want to avoid is, let’s say, saving all your samples for the month in that same file and enable the 10 gigabyte file, it’s just going to slow your software down, it’s going to be difficult to move around. It can be a real pain when it comes to sort of maneuvering these giant data files. So typically, if you’re under a gigabyte, you’re usually in really good shape.
The next thing that you can do is scheduling your antivirus and your Windows updates when you’re not acquiring data, so having an antivirus, especially if you’re connected to the network is probably a requirement for your IT department. And it’s not a bad idea. But we want to make sure that when it’s scanning our files, that we’re not actively acquiring data that can run into issues with data quality, create gaps in our data. So we don’t want that same thing with Windows updates, we certainly don’t want our computer to restart in the middle of an important run.
And last but not least from a maintenance standpoint, disk defragmentation is important. It’s just a way of making all the space in your computer usable and efficiently usable. So again, once a year, once every couple of months when you’re doing that LLC maintenance, when you’re doing your mass spec maintenance, go ahead and let the disk defragmenter run. So if you type in your little search bar down on the bottom of your screen, this defrag will pop up and you could just let it run, it might take quite a while if you haven’t done it. So I’d recommend doing that overnight or something like that.
Slide: 7 best ways to break your LC-MS/MS system #
[16:35] All right, so let’s get into some of the fun things. So we’ve got some examples of some issues. And I took a second and thought about some of the most efficient ways you can break your mass spectrometer. So after all the years on the tech support team and dealing with customers, I’ve seen some pretty creative ways. I’ve done some pretty creative things. So here’s my list of the top seven ways to break your mass spec.
) Not using divert valve, we talked about that. But another interesting one, or really effective one would be using improper buffers, or injecting samples with a lot of salts. So a long time ago, I was working in a lab at a university. And at that university, we had a bunch of HPLCs and just one LCMS in the corner. And the HPLCs were pretty much running all the time just doing UV analysis. And we had a new undergraduate that we had hired in. And they had just started and it was a Friday night. And they didn’t really want to make new mobile phase for the LCMS. We’ve all been there. And they’re so new that they didn’t realize that the mobile phase from the HPLC V system was not the same as the mobile phase from the LCMS system. So they just went ahead, took that bottle, put it on the LCMS and submitted the run for over the weekend. And unfortunately, that mobile phase had a ton of mass spec incompatible buffers in it. And it also had a really high concentration salts. And when we got back in the morning, Monday morning, after a long weekend of running, there was quite a big buildup of salt on the mass spec. And there’s a lot of already corrosion occurring. And we ended up having to replace a ton of the front end to get the system operational again, so an easy mistake. But when you’re building your methods, when you’re going through literature, when you’re trying to really nail down that chromatography, make sure that you’re using LCMS compatible buffers.
) Another really common thing that I see is starting at a really high flow rate of aqueous. So just really high flow rate of water and having a source temperature that’s just a little too low. So what’s happening is you’re essentially not getting that evaporation, and there’s just water running down your curtain plate. Eventually, that water will run down into your source exhaust and clog resource exhaust. And I’ve seen sources flood that way. So you want to make sure when you’re building your methods, that you’re using a temperature that’s appropriate to get your evaporation. And if not that you’re at least using that divert valve. So it’s not going to your mass spec, it’s going to waste when it’s more that high or or high aqueous content.
Slide #
But let’s get into some of the problems. So we come into lab, there’s issues when there’s one issue, there’s many issues, what do we do? And anytime we do any troubleshooting, we’re going to want to try to isolate the issue.
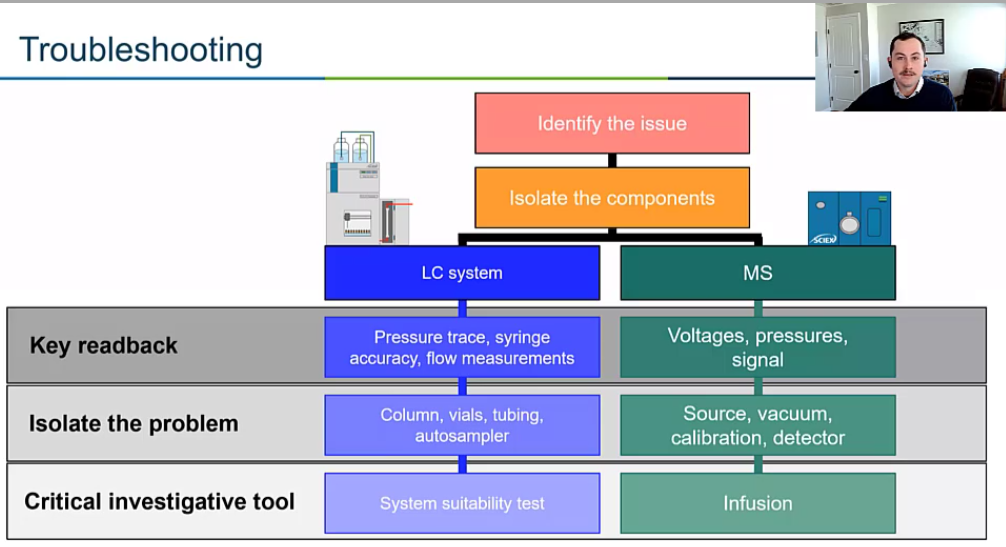 So figure out what where’s the issue coming from? Is it an LC issue? Is it a mass spec issue? The next thing we want to do is designate some key read backs or essentially key variables that we can use to assess the problem whether it’s getting better or worse. This might be intensity. This could be my pressure but it could be just the overall vacuum pressure, for example. So we want to have this sort of key Rebeck that we’ll use to assess whether we’re fixing the problem, or the problem is getting worse.
So figure out what where’s the issue coming from? Is it an LC issue? Is it a mass spec issue? The next thing we want to do is designate some key read backs or essentially key variables that we can use to assess the problem whether it’s getting better or worse. This might be intensity. This could be my pressure but it could be just the overall vacuum pressure, for example. So we want to have this sort of key Rebeck that we’ll use to assess whether we’re fixing the problem, or the problem is getting worse.
And then we want to have some tools. So just two quick examples of tools. But system suitability test is a really great tool for your LC. And then when we’re talking about the mass spec, right, we want to isolate the mass spec. So usually, we’ll be doing some sort of infusion.
Slide: Case #01 #
So let’s start out with an extremely common, but pretty easy problem to fix when you get into the lab.
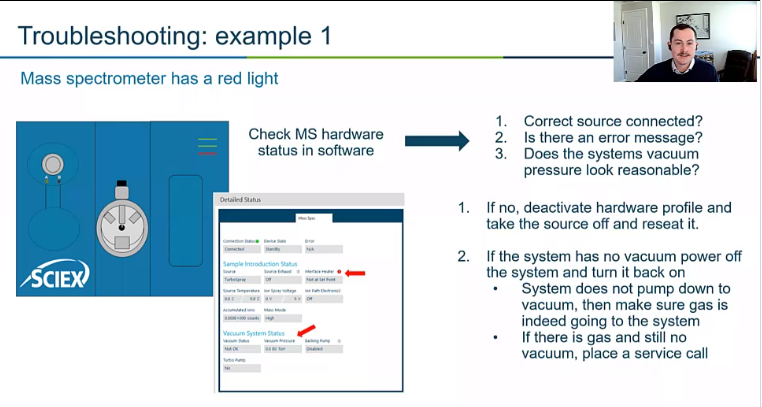 And there’s a red light on our mass spec. So we go into our software, and we notice there’s some some errors here.
And there’s a red light on our mass spec. So we go into our software, and we notice there’s some some errors here.
So the first thing we want to do is say, All right, so the mass spec has an error. Is there something obvious? Did I forget to put the source on? Maybe I didn’t see the source correctly? Are there any error messages that can give me some clues, and also just the vacuum pressure look reasonable?
So you’ll notice the vacuum is quote, not okay. So status, not okay, that’s not a good, it’s not a good status. So we’re going to want to try to figure out why that is occurring. Right. So we’re going to start at the roughing pump. is the roughing pump on? If so we can move on, why isn’t the mass spec pumping down? The most common thing I’ve seen for this is a faulty gas generator. So your mass spec will not pump down to a certain state until that mass spec knows it has nitrogen in there. So if your generator your gas generators off, or there’s some error with it, maybe the flow rate is just way too low, your mass spec won’t even try to pump down. So check your gas generator, if that’s the case, turn back on your gas generator or increase that pressure. And then we’re just going to power cycle or an aspect. Since it’s in this case, not bent, or it’s completely vented, we can just go ahead and hit that power switch. But if it was not, we want to make sure that we hit that little button and slow down that turbo. And if none of those things work, well, then we’re probably going to have to put in a service call.
Slide: Case #02 #
Another sort of really common, but pretty fixable and pretty easily fixable problem with just the general software issues. Again, that computers incredibly important. That’s how we control our instrument. It’s how we control the LC. But there can be communication errors. And I’d say over the years, communication errors end up being the problems you’ll see a at least 80%. I think that’s been a little conservative. But luckily, they’re really easy to fix. So one of the things you can do is just isolate what is the problem is the mass spec not connecting? Or is it the LC. And if it’s the say, in this case, it’s the LC, the next step would just do a power cycle. So shutting down your software, closing the services. And if you don’t have access to the services, just go ahead and restarting your computer. And while you’re doing that, going through just turning the power switch off on your pumps, your auto sampler column oven, your entire LC stack, and leaving it off I typically when I turn off a power switch for a mass spec or an LC, I like to wait 10 minutes before switching it back on again. And when I do switch it back on again, I’m going to wait another minute or so before it tried to connect to it with the software. A lot of times, for example, your auto sampler might do some initialization where it’s trying to home the needle, make sure everything’s working properly. And I don’t want to interrupt it while it’s doing that. So just restarting, doing a power switch or power cycle is usually enough to sort of fix these more common issues.
Slide: Case #03 Shifting retention times example 1 #
[23:55] Let’s move into some exciting sort of not as common issues. So here we have an example of peeks that are just alluding earlier and earlier. So we pretty confident this is an LC issue, right? There’s not really anything from a mass perception perspective that could cause this to happen. So let’s say we run our system suitability test and everything looks fine. Or maybe everything doesn’t look fine. We can use that information to kind of decide which variable might be wrong or what issue we’re having.
Potential causes
So some of the reasons this could be occurring would be there’s an issue with your pump. So maybe one of your pumps isn’t pumping efficiently. Maybe there’s air in the line, but more likely, your column isn’t equilibrated enough. So we talked about it in the method development section quite a bit, but we need that column to be equilibrated to the starting conditions. So we want that column at the starting conditions and the same starting conditions for every injection. So if you maybe shortened your method a little bit, eliminated have some of that set, that a calibration step, this could be an issue you’re having, or just if this is the method you’re building and you’re receiving this. So it’s a really easy problem to fix, all we need to do is on our gradient, add a little bit more time to that equilibration step. So when we come back down from that high organic to our starting conditions, add a minute or two, if you want, you can calculate exactly how much time you need to add, typically, three column volumes is enough to equilibrate your column. But if not, add 30 seconds to a minute, and see if this problem persists.
Slide: Case #03 Shifting retention times example 2 #
So now another issue with retention time would be just your peaks moving all around. So this is something that’s happened to me a bunch, where you just see one injection, you know, your retention time is seven and a quarter, and then you inject immediately after that. And now it’s eight minutes, and it’s just kind of bouncing around randomly. So this can be a pretty frustrating problem, especially if you have retention on Windows. And these shifts caused them to be outside those windows, so essentially not seeing that peek.
Potential causes
More likely than not, this is just caused by some air in your lines. So we talked about purging, I always like to purge in the morning, it’s really easy to do. Just go ahead purge your A line and your B line and your rinse line, if you have one and take the two minutes and eliminate this problem. That being said, if that doesn’t fix your issue, there are a couple places that air can kind of hide in your LC system. And the most common one I see is the solvent filters. So that little filter on the bottom of your Eluant line that’s in your bottle. A lot of times they’re metal, sometimes if they’re brand new air can kind of get stuck in there. And it can be pretty difficult to remove. So if that’s the case, you can go ahead and take them off, put them in methanol or your mobile phase itself, and just sonicator them quickly. And that will get rid of a lot of the air, you put them right back on, and you should be good to go.
Slide: Case #03 Shifting retention times example 3 #
But let’s say that is happening. So you know, Monday morning, we come in, and we’ve got this nice peak, it’s elluting right in our retention time window, and then all of a sudden on Tuesday, we don’t see that peak anymore.
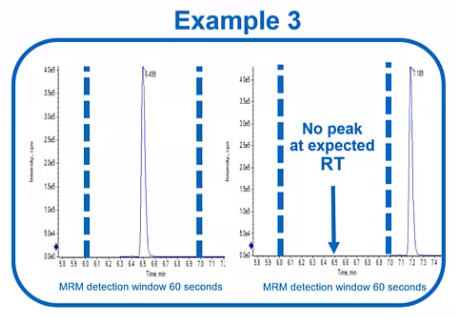 So the first thing we need to figure out is, is this a mass spec issue? So has their sensitivity somehow completely disappeared, and we just can’t see that peak? Or is it an LC issue? is that peak eluting outside of our retention time window? So typically, the easiest way to sort of address this, would be to go ahead and run an unscheduled method. So if you remember from the method development section, when we talk about scheduled methods, we’re telling the instrument, hey, look for this peak at this retention time, versus an unscheduled method where it’s constantly looking for that peak. Now, if you have a lot of analytes, the data quality might not be great. But you can go ahead and unscheduled, just the peak that you have an issue with or just unscheduled them all. We’re not using this for quantitation. We’re not super worried. If there’s only three or four points across our peak at this point, we just want to know is that peak eluting? Or is there another issue that would be more mass spec related. And if we do see a peak, there are a few things we can do to try to address this. So let’s say our peak is just outside a retention window.
So the first thing we need to figure out is, is this a mass spec issue? So has their sensitivity somehow completely disappeared, and we just can’t see that peak? Or is it an LC issue? is that peak eluting outside of our retention time window? So typically, the easiest way to sort of address this, would be to go ahead and run an unscheduled method. So if you remember from the method development section, when we talk about scheduled methods, we’re telling the instrument, hey, look for this peak at this retention time, versus an unscheduled method where it’s constantly looking for that peak. Now, if you have a lot of analytes, the data quality might not be great. But you can go ahead and unscheduled, just the peak that you have an issue with or just unscheduled them all. We’re not using this for quantitation. We’re not super worried. If there’s only three or four points across our peak at this point, we just want to know is that peak eluting? Or is there another issue that would be more mass spec related. And if we do see a peak, there are a few things we can do to try to address this. So let’s say our peak is just outside a retention window.
Potential causes
the easiest thing to do would be to make some new mobile phase, maybe we’ve let our mobile phase sit for quite a while. We’re using formic acid, it’s volatile, and it’s sat too long. And now we have this retention time shift.
Another really easy thing to do would just be to try a new batch of buffer. So let’s say again, we’re using formic acid, maybe that formic acid super old, maybe it’s been left out on the counter overnight without a cap. Maybe the quality of it just isn’t very good. Try a new batch is usually a really easy solution to see, hey, does that help.
And then finally, just trying a new column see if that column has sort of run it’s life. And maybe a new one will address this problem. You’ll notice that a lot of the LC and retention time issues in particular, often can be attributed to columns. So the easiest thing or the best thing, I think, in my opinion is to go ahead, purchase an extra column, keep it in your drawer, even if you’re not using it. It’s a really fast way to assess is it the column or is it the LC? Since that’s going to be the area you kind of focus on first.
Slide: Case #04 High Background #
[29:45] But let’s say we have this issue, let’s say we have current data and historical data.
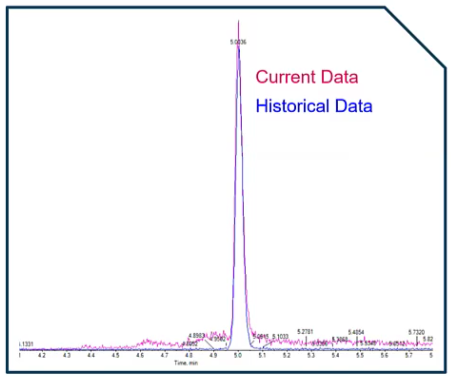 So the current data in pink and historical data in blue, and if we look at this would say hey, everything looks pretty good. Our peak heights are very similar. I have no issue if this isn’t a low point in my career if I might not even notice what’s going on.
So the current data in pink and historical data in blue, and if we look at this would say hey, everything looks pretty good. Our peak heights are very similar. I have no issue if this isn’t a low point in my career if I might not even notice what’s going on.
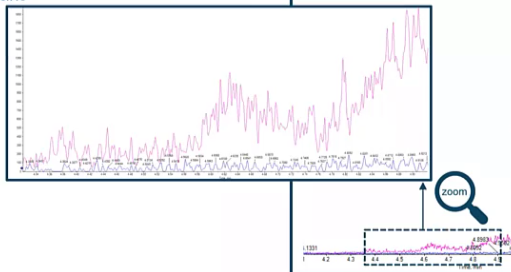 But if we zoom in to our background, what we see is, there’s been a really big increase in our background. So we’re starting to see a lot of background, about 1500 CPS, compared to our traditional background, which was around 100 CPS, so a 15x increase in background. Now, this might not be a problem right away. Maybe if this is the low point of our curve, it’s not a problem for a long time. But if it’s not, and we wanted to sort of go a bit lower or take a look at sort of our true LOD on a system, we’d likely have a pretty hard time. Because in the past, if our peak was 1500, CPSI, for example, that’s a well defined peak from our background, based on historical data. But in our current data set, you would not really notice that peak was there. So you could lose that information. And we don’t want that.
But if we zoom in to our background, what we see is, there’s been a really big increase in our background. So we’re starting to see a lot of background, about 1500 CPS, compared to our traditional background, which was around 100 CPS, so a 15x increase in background. Now, this might not be a problem right away. Maybe if this is the low point of our curve, it’s not a problem for a long time. But if it’s not, and we wanted to sort of go a bit lower or take a look at sort of our true LOD on a system, we’d likely have a pretty hard time. Because in the past, if our peak was 1500, CPSI, for example, that’s a well defined peak from our background, based on historical data. But in our current data set, you would not really notice that peak was there. So you could lose that information. And we don’t want that.
Potential causes
So some of the things that can be causing this, it could be your column again, so you could have some kind of aging, contamination,
the mobile phases is probably a really good place to start. So try a new batch of mobile phase, make sure you’re washing your bottles three times before you do it. And then let’s say you’re using one vendor for your aqueous, try another vendors, see if that helps, see if that removes some of your background. And if it does, great. If not, then you can go after maybe the column or the buffer solution. But typically, when we see this, it’s going to be mobile phase related.
Slide: Case #05 Peak Shape Deterioration #
Another issue that we run into quite a bit would be we had really great peak symmetry, everything was looking great.
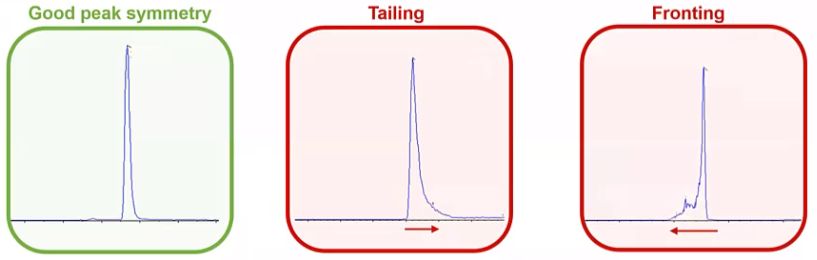 And now we have tailing, perhaps, or we even have fronting. So what can be causing that?
And now we have tailing, perhaps, or we even have fronting. So what can be causing that?
Potential causes
Again, it could be a column issue. Another thing that can occur is if you’ve changed your sample composition, so let’s say in your earlier method, you were injecting 50%, water, 50% methanol, and now you’ve increased it to 60% methanol or 70% methanol, that can have a big effect on your peak shape. So that could be a cause.
But if nothing’s changed, the most likely culprit is just a leak. So recheck your fittings, go ahead and make sure that your confidence are tight and secure, you don’t have a leak anywhere, these leaks would be really hard to find they’re likely very, very small. So what I would typically do is just disconnect the column completely, and then reconnect it again, make sure that the fittings aren’t, for example, damaged in some way.
Slide: Case #06 Quantification #
[32:48] Number six. So here we have a quantification issue. And this is one that we see a lot at Sciex, and it’s, it’s just a silly one that we can fix really quickly. So we’ve got this beautiful curve, we’re very happy with it. And then we end up when we go to our next data file the next day.
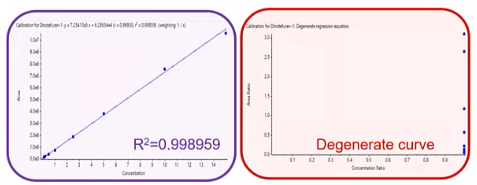 But this degenerative curve, so we see, oh, it’s just a straight line, and not linear in the way we want. It’s just perfectly horizontal. So what could be causing that we take a look at our peak areas, and we say, hey, our areas look good. Our internal standard areas look good, they’re consistent.
But this degenerative curve, so we see, oh, it’s just a straight line, and not linear in the way we want. It’s just perfectly horizontal. So what could be causing that we take a look at our peak areas, and we say, hey, our areas look good. Our internal standard areas look good, they’re consistent.
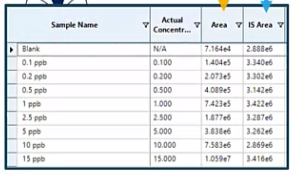 Why is this occurring? And more than likely, in fact, almost definitely, what happened is you may have accidentally entered in concentrations for your internal standard concentration. So typically, when we use an internal standard, as we discussed last time, they’re added at the same amount. Whereas here, accidentally, you’ve entered them in as if they were occurring themselves.
Why is this occurring? And more than likely, in fact, almost definitely, what happened is you may have accidentally entered in concentrations for your internal standard concentration. So typically, when we use an internal standard, as we discussed last time, they’re added at the same amount. Whereas here, accidentally, you’ve entered them in as if they were occurring themselves.
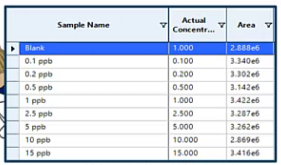 So when we’re doing that ratio, if you remember from last time, we’re we’re taking the area of the internal standard over the area of your analyte. We’re ending up with this perfectly horizontal line. In our software, for example, there’s an option to apply concentrations to all and if you don’t have your internal standards listed as internal standards, when you do this, it’s going to apply just your standard concentrations. Super easy fix though, all you’ve got to do is go into your software and enter the true concentration of an internal standard. But I figured I’d mention it because I’ve seen it a lot. And it’s just an easy thing to happen. But it might not be super obvious the first time it happens to you.
So when we’re doing that ratio, if you remember from last time, we’re we’re taking the area of the internal standard over the area of your analyte. We’re ending up with this perfectly horizontal line. In our software, for example, there’s an option to apply concentrations to all and if you don’t have your internal standards listed as internal standards, when you do this, it’s going to apply just your standard concentrations. Super easy fix though, all you’ve got to do is go into your software and enter the true concentration of an internal standard. But I figured I’d mention it because I’ve seen it a lot. And it’s just an easy thing to happen. But it might not be super obvious the first time it happens to you.
Slide: Case #06 Quantification #
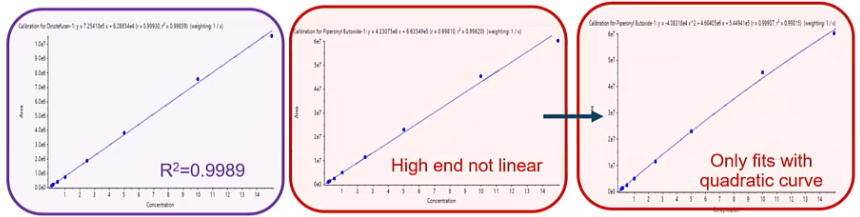
[34:38] Here’s a scenario where we have a linear curve, everything’s looking great. But over time a curve becomes nonlinear. In this case, it’s more quadratic.
Potential Causes
So what could be causing this? And that can be a few different things. It’s gonna depend a little bit based on how you built your method and when you build your method. One of the times I’ve seen this or several of the times a few of the times I’ve seen this have been when a method was built on an instrument that wasn’t operating at its full sensitivity. So let’s say, you know, your instrument was sort of dirty, or maybe it was your detector wasn’t fully optimized correctly. So your sensitivity wasn’t as great as it could have been. So when you built your curve, initially, it was linear. And then your engineer yourself, went in, optimized, clean, etc. And now you’re seeing this quadratic curve. If that’s the case, there’s no big issue, we can just adjust the method to sort of account for this. And the easiest way to account for this, regardless of the cause would be to go ahead and either inject less. That way, you don’t have to change any of your sample preparation procedures, or your SOP or anything like that. And the other option would be just to use a slightly less concentrated solution. So you can decrease your your upper LOQ, will depend a little bit based on kind of your goals and what you’re trying to accomplish which way you choose. But you have a couple of options there to sort of eliminate this issue.
Slide: Case #07 Low Sensitivity #
And then the classic one, low sensitivity. So this is very clearly a mass spec problem. We’ve determined the LC is operating completely, normally, we’ve tried new mobile phase, etc, we can isolate it to our mass spec.
Potential Causes
And the easiest way to do this is with an infusion. So we can take a tuning solution, so one of the ones provided with your instrument, or we can simply use a compound that we know the relative sensitivity what it should be, and infuse it. And if we noticed that, hey, there is this drop off in sensitivity. It’s simply not as sensitive as it used to be. The most likely culprit is just a dirty mess spec. And that could be your probe, that could be your curtain plate, that could be your Q-jet. Hopefully not too much further than that. So the easiest thing to do would be to go ahead and clean. if you’re comfortable doing the front end cleaning. So meaning that you’ve removed your curtain plate or orifice plate, you’ve taken out your cue check, you can do that. So typically, when I go ahead and clean a Q-jet, I’ll go ahead and take it out, put it in a beaker of water, sonicated for 10 or so minutes, then put it in a beaker of methanol sonicated for 10 or so minutes before putting it back in. That’s a really easy, quick way to sort of clean your Q-jet. If it’s just a little bit dirty. If it’s more dirty than that, you might have to do a little bit of disassembly. Or if you’re uncomfortable doing those types of front end cleanings, you can go ahead and just schedule a service call. And they can get it sorted for you pretty quickly. But having that sort of tool, that infusion tool and a known solution, whether it be your tuning solution is really key here, because that’s going to identify that yes, the problem was the mess back. And more than likely, it just needs to clean.
Slide: Case #08 HPLC Clog #
[38:10] What about clogs, we see them all the time, especially for dealing with some nasty samples. So we’ve got this higher pressure than we’d expect. Where maybe our pumps are maxing out and stopping. Because the pressure is so high. How do we address this.
Potential Causes
So it’s likely a clog. The easiest way to sort of address and troubleshoot a clog would be to go through and systematically remove pieces of tubing. Usually, you’d want to start at the mass spec, remove the probe has the pressure gone down, remove the piece of tubing from the diverter valve to the probe has the pressure gone down, and so on so forth, and just move your way up the line. And as you move your way up the line, eventually when you remove a piece of tubing, that pressure will drop. And when it does, you’ve isolated the problem, you can go ahead and replace that piece of tubing and see if your pressure is in a normal range or if there’s more tubing that needs replaced.
Slide: Case #09 Missing data #
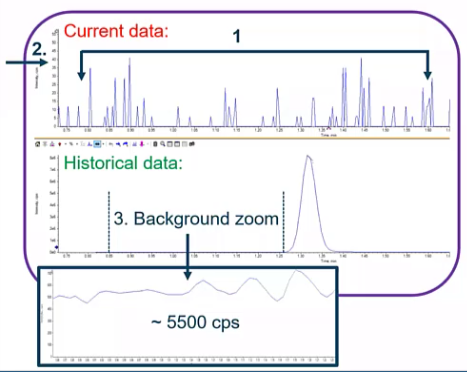
[39:07] But pretty simple. Compared to something like missing data, so missing data is one of the more frustrating things you’ve submitted around, everything looks good. And they come in and you see, hey, there’s only this electrical noise. So there’s no background, there’s no peak. If I look at my background, previously, there was sort of this around 5500 CPS background and now there’s just nothing.
 Potential Causes
I’ve definitely been here before. And when it’s a new method, I may have forgotten to program my Diverter Valve properly. So instead of going to my mass spec, it was always going to my waist. But if it’s not a new method and nothing has changed, you’d likely have a leak.
Potential Causes
I’ve definitely been here before. And when it’s a new method, I may have forgotten to program my Diverter Valve properly. So instead of going to my mass spec, it was always going to my waist. But if it’s not a new method and nothing has changed, you’d likely have a leak.
 More than likely it’s probably in your column oven. So go ahead, pop up in the column oven, ensure all your fittings are tight. Check your source make sure that you’ve got added that piece of tubing to your actual probe. But more than likely, there’s a leak somewhere if all of a sudden you see this issue after switching columns, for example.
More than likely it’s probably in your column oven. So go ahead, pop up in the column oven, ensure all your fittings are tight. Check your source make sure that you’ve got added that piece of tubing to your actual probe. But more than likely, there’s a leak somewhere if all of a sudden you see this issue after switching columns, for example.
Slide: Case #10 carryover from flow path #
And for number 10. So this is actually one that was requested. Last time we asked some of the questions. And this was about carryover. So we had run a high standard, and now we’ve run a blank, and we have some carryover that we need to address.
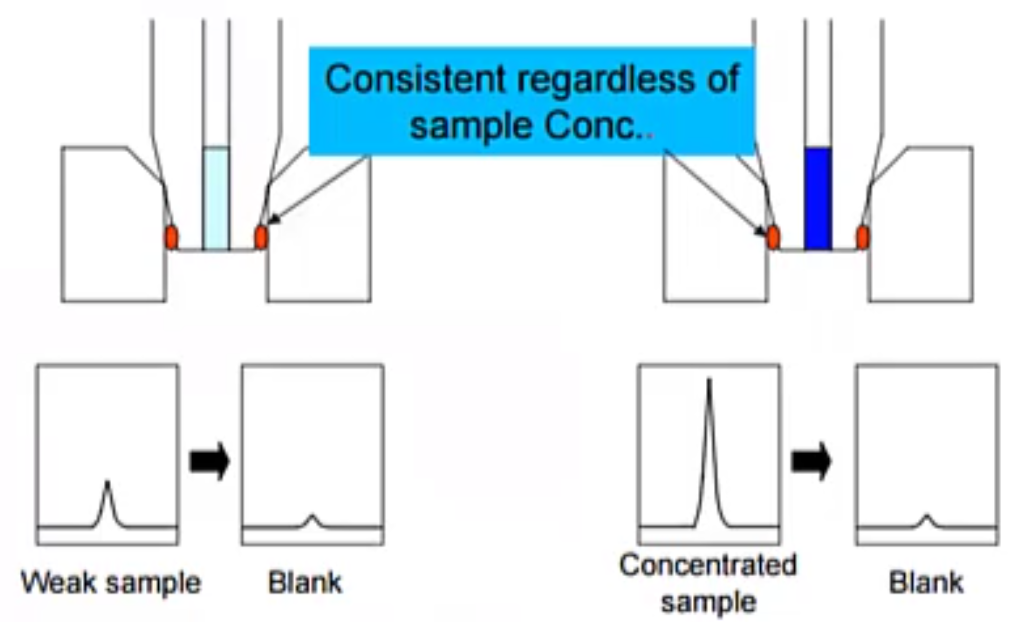 Well, there are two main sources for carryover and they present pretty differently. If it’s coming from the autosampler, like in this case, where we have just some consistent contact with the standard and our autosampler Needle, what we’ll see in our sample is just this continuous peak. So it doesn’t matter how much we inject, the peak is always the same. So if we go ahead and inject 10 microliters, our peak and our blank is going to be the same height. If we inject 20 microliters, the peak in our blank is still the same height. So that’s telling us that it’s just from contact, a couple of ways to address this would be go ahead and inject less or reduce your standard concentration volume.
Well, there are two main sources for carryover and they present pretty differently. If it’s coming from the autosampler, like in this case, where we have just some consistent contact with the standard and our autosampler Needle, what we’ll see in our sample is just this continuous peak. So it doesn’t matter how much we inject, the peak is always the same. So if we go ahead and inject 10 microliters, our peak and our blank is going to be the same height. If we inject 20 microliters, the peak in our blank is still the same height. So that’s telling us that it’s just from contact, a couple of ways to address this would be go ahead and inject less or reduce your standard concentration volume.
Another option is if you’ve set your protrusion depth very deep, you can back that off a little bit. So go up a little bit and make sure that you’re not completely submerging the needle, that can sometimes help as well.
Slide: Case #10 carryover from column #
[41:23] If that’s not the case, and that contamination is coming from your column, well, then there’s another solution for you.
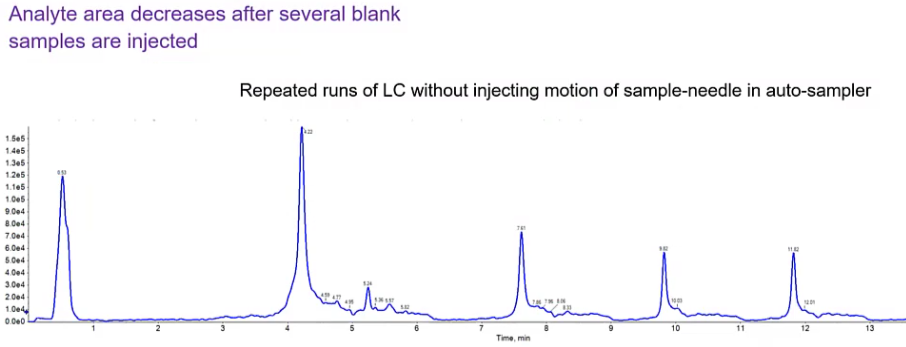 And the easiest way to kind of determine between the two are to try to figure out whether it’s from your column, or it’s from your injector, is to do these repeated gradients. So this is a single run, where I’ve done the same gradient four times.
And the easiest way to kind of determine between the two are to try to figure out whether it’s from your column, or it’s from your injector, is to do these repeated gradients. So this is a single run, where I’ve done the same gradient four times.
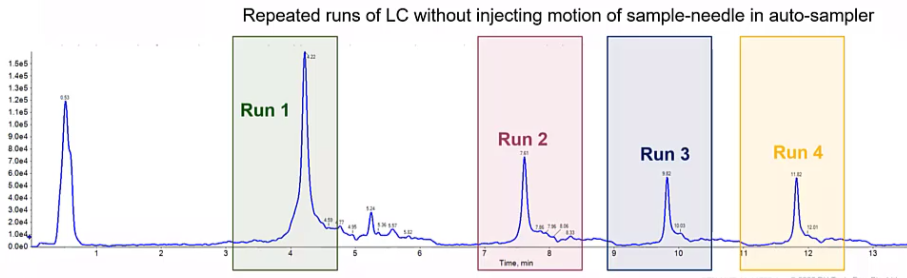 So what I see is that I have a contamination and run to it’s still there, it’s decreasing over time. But that contamination is still present. So what this is telling me is that it’s not from the auto sampler, because my auto sampler isn’t doing anything. So it hasn’t injected anything. It hasn’t it’s valves haven’t changed, it’s just been sitting idle. And instead, we’re just flowing through right through a column. So likely this is a column. More than likely, it’s maybe we don’t have an long enough whole time for organic. So we talked about sort of that column equilibration time here, just increasing that column whole time or that organic hold time, may help. So again, typically three column volumes. But if we increase that a minute or two, maybe we’ll see this determination decrease. The other option, obviously, is trying another column.
So what I see is that I have a contamination and run to it’s still there, it’s decreasing over time. But that contamination is still present. So what this is telling me is that it’s not from the auto sampler, because my auto sampler isn’t doing anything. So it hasn’t injected anything. It hasn’t it’s valves haven’t changed, it’s just been sitting idle. And instead, we’re just flowing through right through a column. So likely this is a column. More than likely, it’s maybe we don’t have an long enough whole time for organic. So we talked about sort of that column equilibration time here, just increasing that column whole time or that organic hold time, may help. So again, typically three column volumes. But if we increase that a minute or two, maybe we’ll see this determination decrease. The other option, obviously, is trying another column.
Slide: Case #11 Contamination #
And last but not least, this is one of my favorite sort of case studies that happened to us.
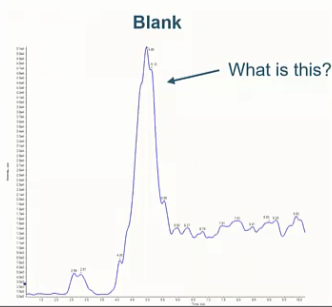 So this is contamination, contamination is a big issue. In in all labs, right, we’re worried about it. And in this lab in particular, and they’re blanks, they’re having a contamination issue, we determined it wasn’t carryover, we tried running a new blank. That didn’t work, we tried to different methanol source, like before it maybe it’s coming from our mobile phase, that also didn’t work. In the past, I’ve seen dirty pipettes cause things like this. So we just removed the pipette completely or tried a different pipette. And that was the case. And then finally, we just tried a new column. Again, we’re still seeing this background peek. So maybe it’s something that’s just contaminated our LC, and we need to flush out so we flushed the system for a few hours with methanol and formic acid. And we saw no decrease in this peak height.
So this is contamination, contamination is a big issue. In in all labs, right, we’re worried about it. And in this lab in particular, and they’re blanks, they’re having a contamination issue, we determined it wasn’t carryover, we tried running a new blank. That didn’t work, we tried to different methanol source, like before it maybe it’s coming from our mobile phase, that also didn’t work. In the past, I’ve seen dirty pipettes cause things like this. So we just removed the pipette completely or tried a different pipette. And that was the case. And then finally, we just tried a new column. Again, we’re still seeing this background peek. So maybe it’s something that’s just contaminated our LC, and we need to flush out so we flushed the system for a few hours with methanol and formic acid. And we saw no decrease in this peak height.
 So then we started thinking, Well, maybe it’s not from our mass spec, maybe it’s from our surroundings, right? So we’ve kind of figured out that it’s not really from the LC system, it’s really not mass spec related. It doesn’t look like it’s a file, sample prep, etc. What about our environment. So we went around swabbing, which is taking a chem wipe with a little bit of methanol on it, it’s swabbing different surfaces around the lab. And in the prep room, the peak was a little bigger, but it was pretty much the same. outside the lab, we noticed, okay, not a whole ton of difference. And then compared to our blank, really, we’re just seeing the same thing. So we haven’t really identified the source yet. But talking to the customer, and they mentioned that they had a new air conditioner installed in that lab about a month ago. And, you know, we kind of started thinking, we’re like, oh, that’s exactly when this problem started. So we went ahead and swapped that air conditioner, and got a big peak for this compound. So what had happened where they’re using some similar compound wasn’t the exact same compound, but it was sharing an MRM transition and causing this really high background peak that was causing them not to be able to run that assay.
So then we started thinking, Well, maybe it’s not from our mass spec, maybe it’s from our surroundings, right? So we’ve kind of figured out that it’s not really from the LC system, it’s really not mass spec related. It doesn’t look like it’s a file, sample prep, etc. What about our environment. So we went around swabbing, which is taking a chem wipe with a little bit of methanol on it, it’s swabbing different surfaces around the lab. And in the prep room, the peak was a little bigger, but it was pretty much the same. outside the lab, we noticed, okay, not a whole ton of difference. And then compared to our blank, really, we’re just seeing the same thing. So we haven’t really identified the source yet. But talking to the customer, and they mentioned that they had a new air conditioner installed in that lab about a month ago. And, you know, we kind of started thinking, we’re like, oh, that’s exactly when this problem started. So we went ahead and swapped that air conditioner, and got a big peak for this compound. So what had happened where they’re using some similar compound wasn’t the exact same compound, but it was sharing an MRM transition and causing this really high background peak that was causing them not to be able to run that assay.
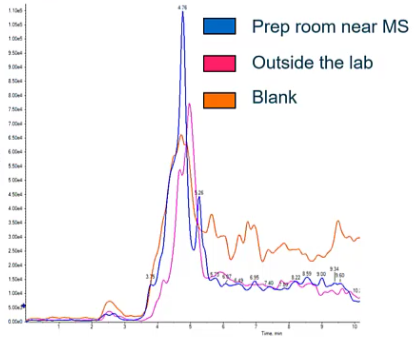
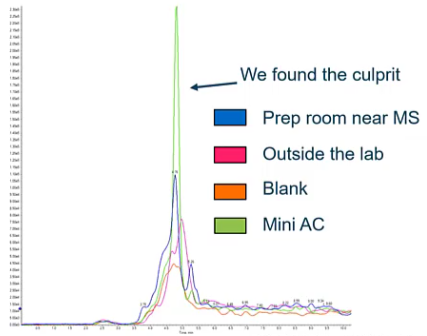 So when we figured this out, we took the AC out of the room. They ended up cleaning it and reassembling it and their background went back down. But it’s something to think about when we think about contamination, it maybe from the LLC, and maybe even mass spec, but we also have to remember that it could also be from the environment.
[Note] Visualization would be a big tool to catch the date when it happened.
So when we figured this out, we took the AC out of the room. They ended up cleaning it and reassembling it and their background went back down. But it’s something to think about when we think about contamination, it maybe from the LLC, and maybe even mass spec, but we also have to remember that it could also be from the environment.
[Note] Visualization would be a big tool to catch the date when it happened.
Slide #
Regardless, in all these cases, troubleshooting is an important part of what we do day to day. And the one thing I’ll say is, the more practice you get with troubleshooting, the better you become. So if you’re a scientist who’s just starting out, Shadow your senior scientist, because they’ve probably seen the same scenario before, and they can help you get to the bottom of the problem a little bit faster. And if your senior scientist in includes some of those more junior scientists into your, into your troubleshooting process, it’s going to help them out and it’s going to help you out in the long run when you don’t have to fix their problems as much. Regardless, though, it’s all about isolation. So isolate where the problem is, is it a mass spec problem, computer problem, LC problem is it my environment, that’s going to be really critical for getting to the bottom of your problem as quickly as possible.
Slide #
If you’re interested in more of the maintenance courses, say you really want to learn how to do a front end cleaning, I encourage you to check out sites now they have great videos that go through step by step from removing the Q-jet, disassembling it, cleaning the lens, all of that can be found here. So go ahead, sign up. Those courses, I believe are free. So they can be a really great resource.
And with that, I just want to remind you that you can check out any of these videos on demand and continue to watch this series. And let us know in the future kind of some of the things you’re interested in, because we really want to provide you with tools and solutions for the problems that you’re facing in your lab.
Q&A #
Thank you for that insightful presentation, curl around maintenance and troubleshooting. I hope you learned a couple of tips and tricks to keep your system in tip top shape. And now we’ll open the floor for questions.
Q#1
So the first question is around the probe. So you mentioned Karl, the need for cleaning the probe on the ion source? Can you Sonicate the entire probe? And if that’s not the cleaning mechanism, what do you recommend as far as a cleaning protocol for your ion source probe?
Yeah, great question. So you can suffocate the entire probe, or I wouldn’t recommend it. But what you can do is take out that electrode. So when you check your protrusion, right, that electrode that sticks out at the bottom of your probe, you can remove that and sonicated that completely. So that’s okay to be completely submerged. If you wanted to, you could clean the housing of your pro, but it really shouldn’t affect with like clogs, or anything like that, since your fluid is going through that electrode, right. So I wouldn’t worry too much about it. Usually, you can just clean the probe with the kim wipe, and just wipe it down quickly.
Q#2
Excellent. Okay, on to the next question, which is about cleaning mobile phase bottle. So you mentioned a few times that somebody should clean their mobile phase bottles, what solvents, or solution mixtures should be used to clean the mobile phase bottles for the LC system?
Yeah, great question. So some sort of organic is typically sufficient. So something like I see aectonitrile, and methanol, the big thing we want to do is make sure that we’re washing them multiple times. So when we go ahead and clean with methanol, for example, we’re going to rinse once dispose of that solution, rinse again with methanol, dispose of that solution, and so on, so forth. Some labs do use something like alconox or other class where cleaners, I’ve always kind of steered away from that a little bit, because it can cause issues if you don’t organize them properly. So if you have a really, really dirty mobile phase bottle, maybe maybe you need to do that. But most of the time, these mobile phase bottles, right, they’re relatively clean. So triple rinsing was something like methanol or acetonitrile is usually is sufficient.
Q#3
Excellent. Next question was around mobile phase filtration. You didn’t mention mobile phase filtration, do you feel the need to filter your mobile phases? And if so why?
Yeah, great question. So no, I really never do any mobile phase filtration. In general, I typically find that that adds more contaminants than it removes, at least in my case. Instead, what I always recommend is purchasing those high grade solvents, those LCMS grade solvents, because that’s essentially what the solvent vendor has done. It’s filter it and they have very specific procedures and QA QC requirements for that solvent. So they just do a better job than me so I don’t do it in house. I purchase it from them.
Q#4
Excellent. The next question is around system suitability. You talked about the need for system suitability mixes. do you have any suggestions for test compounds that you might want to put into mixes? And and if not, maybe a strategy for how based on their application or need, they might select a system suitability mix?
Yeah. So one of the easiest ways to selected selected system suitability mix is if you have an instrument, let’s say your mass spec is constantly running pesticides. That’s really all it does. is it runs this pesticide mix, well, then you have a great historical data of what you know what that mix looks like over time. So that kind of is your system suitability next. The other option if that’s not the case, so let’s say you’re maybe an acedamic lab, or maybe this mass spec is being used for multiple different assays, choosing something really simple like [???] if you’re looking at positive mode or a vertical, if it’s negative mode, something that is really accessible. And ionization really well is usually what I would suggest, and you don’t need a ton of different compounds. Just having the single compound can give you a lot of information.
Q#5
Excellent. Okay. The next question is around mobile phase flow rates. What mobile phase flow rate is too high? Because you talked about too high and flooding the source? How do you determine what is too high?
Yeah, so it’s going to depend based on your source temperature. So if you’re running at a very low source temperature, let’s say 300 mils per minute, or I’m sorry, 300 degrees Celsius. And then you have a flow rate of A mil per minute, and you’re pumping just completely aqueous solvent, it’s probably going to not evaporate and just continue to reach your curtain plate. Versus if it was a mil per minute and was methanol, it would have every little bit easier, right? So there’s a little couple of variables at play. Typically, though, based on our analytical columns that we use for LCMS, I don’t see flow rates really ever above 1.2 mils, and that’s very, very high. Usually, one mil is about the target and range for flow rates.
Q#6
Excellent. Okay. The next question is around blank injections. Do you recommend a blank injection at the end of your batch? And where else do you recommend running blank injections throughout your batch for maximum data quality?
Absolutely. So certainly, I’d say the most important place to run a blank is after your highest standard. So you’ve run your curve, then go ahead and run a blank and see if you have any carryover there. The other thing I like to do, and I think we talked about it last time, it’s sort of sandwich my samples. So I’ll run, let’s say 10 samples, run a QC and then run a blank. So that Qc is going to go ahead and tell me my calibration period still great. But the blank is going to let me know if I have carryover issues. Maybe I ran a really hot sample, and I’m still seeing some caffeine in my next run. So running them sort of in that sandwich, maybe every 10 to 20 samples is usually a good range to ensure your data quality is very high.
Q#7
Excellent. And the next question is also around solvents. It’s using DMSO, as far as a solvent for the sample itself, not as far as mobile phases. Do you recommend using the DMSO as a solvent for the samples? Or do you recommend maybe drying down and reconstituting in more mass spec friendly solvents for samples?
If you can, reconstitution is probably your best bet, especially if you’re going to be running a lot of samples. If it’s something that you’re sort of doing some exploration with, and you’re injecting just a small amount, it can be okay, but I really avoid it as much as possible. Typically, you know, I try to shoot my system isn’t as nice as I can.
Q#8
Excellent. The next question is around multiresidue methods, right? A lot of our customers use the mass spectrometry systems to look at a lot of different analytes in one injection. What’s your first line of defense if you only see a problem with maybe one or two of the analyte? And that methodology and not a whole methodology?
Yeah. So that’s something that actually happens to us quite a bit in our cannabis method, for example, and customers will say, you know, hey, I’m looking for [???]. My [???] sensitivity has really dropped off, but many other analyzer are totally fine. And a lot of times that can be attributed to our modifiers. So if you’ve made in this case, they’re looking at ammonium anions but only a handful of them are actually ammonium anions. So they only see this like loss of sensitivity with their [???]. So the easiest solution or the solution there is likely that their modifier is just a little old, maybe their their mobile phase is old, so they can just try to remake some new one or or if that’s not the case, go ahead, purchase some new ammonium cases ammonium formate some new ammonium formate and try that and see if that works.
Thank you so much for joining us today and for providing those insightful questions. Again, a big thanks to Karl for providing the great presentation around maintenance and troubleshooting. We hope you enjoyed this session of LC Ms. Ms. 101. And we look forward to seeing you at another Sykes event. Have a great day. Stay safe, stay healthy, and we look forward to seeing you soon bye for now